Sentieon
Sentieon 中文手册
Sentieon 中文手册(上册)
Sentieon 中文手册(下册)
Sentieon 软件应用教程
Sentieon | 联合Dell、AMD开发基因组大数据分析加速方案
Sentieon | 应用教程: 利用Sentieon Python API引擎为自研算法加速
Sentieon | 应用教程: 关于读段组的建议
Sentieon | 应用教程: 使用DNAscope对HiFi长读长数据进行胚系变异检测分析
Sentieon | 应用教程: TNscope® 使用机器学习模型进行有匹配正常样本的体细胞变异发现
Sentieon | 应用教程: CCDG使用Sentieon®的功能等效流程
Sentieon | 应用教程: 利用共识功能去除PCR重复
Sentieon | 应用教程: 适用于PacBio HiFi和Oxford Nanopore长读长测序数据的结构变异检测
Sentieon | 应用教程: 使用 Sentieon进行大型基因组重测序分析
Sentieon | 应用教程: 体细胞SNP/Indel变异检测
Sentieon | 应用教程: DNAscope使用机器学习模型进行胚系变异调用
Sentieon | 应用教程: 唯一分子标识符(UMI)
Sentieon | 应用教程: Sentieon分布模式
Sentieon | 应用教程:使用CNVscope进行CNV检测分析
Sentieon发布核心家系(trio)基因分析最佳实践方案
Sentieon推出Segdup-caller:针对片段重复区域的专用精准变异检测工具
Sentieon-cli | DNAscope WES 流程单条命令版本详解
Sentieon | DNAscope 核心家系(trio) WES 分析全流程详解
Sentieon | DNAscope Hybrid长短读长混合分析流程详解及评测
Sentieon | 基于Illumina WGS数据的高精度、低成本的泛基因组分析方案
Sentieon软件版本更新
Sentieon | 发布V202503.01版本
Sentieon | 发布V202503.02版本
Sentieon | 发布V202503.03版本
Sentieon软件快速入门指南
Sentieon 软件模块总述
Sentieon 特色流程 - DNAscope
Sentieon | DNAscope Illumina 流程
sentieon | DNAscope Complete Genomics 流程
Sentieon | DNAscope LongRead PacBio 流程
Sentieon | DNAscope Ultima Genomics 流程
Sentieon | DNAscope Element Bio 流程
Sentieon | DNAscope LongRead Nanopore 流程
Sentieon混合分析流程 - DNAscope Hybrid
Sentieon推出混合型短读长和长读长变异检测DNAscope Hybrid流程(上)
Sentieon推出混合型短读长和长读长变异检测DNAscope Hybrid流程(下)
毅硕Sentieon | 泛基因组分析流程详解
毅硕Sentieon | RNA-seq 变异检测全流程详解
毅硕Sentieon | 物种全基因组(WGS)分析流程
毅硕Sentieon | 植物全基因组(WGS)分析流程
毅硕Sentieon | 小麦(Triticum aestivum)全基因组WGS分析流程
毅硕Sentieon | 水稻(Oryza sativa)全基因组WGS分析流程
毅硕Sentieon | 拟南芥(Arabidopsis thaliana)全基因组WGS分析流程
毅硕Sentieon | 马铃薯(Solanum tuberosum)全基因组WGS分析流程
毅硕Sentieon | 巨桉(Eucalyptus grandis)全基因组WGS分析流程
毅硕Sentieon | 向日葵(Helianthus annuus)全基因组WGS分析流程
毅硕Sentieon | 野草莓(Fragaria vesca)全基因组WGS分析流程
毅硕Sentieon | 银杏(Ginkgo biloba)全基因组WGS分析流程
毅硕Sentieon | 大豆(Glycine max)全基因组WGS分析流程
毅硕Sentieon | 陆地棉(Gossypium hirsutum)全基因组WGS分析流程
毅硕Sentieon | 胡桃(Juglans regia)全基因组WGS分析流程
毅硕Sentieon | 苹果(Malus domestica)全基因组WGS分析流程
毅硕Sentieon | 香蕉(musa acuminata)全基因组WGS分析流程
毅硕Sentieon | 罂粟(Papaver somniferum)全基因组WGS分析流程
毅硕Sentieon | 开心果(Pistacia vera)全基因组WGS分析流程
毅硕Sentieon | 毛果杨(Populus trichocarpa)全基因组WGS分析流程
毅硕Sentieon | 动物全基因组(WGS)分析流程
毅硕Sentieon | 猪(sus scrofa)全基因组WGS分析流程
毅硕Sentieon | 鸡(Gallus gallus)全基因组WGS分析流程
毅硕Sentieon | 家鼠(Mus musculus)全基因组WGS分析流程
毅硕Sentieon | 家犬(canis lupus familiaris)全基因组WGS分析流程
毅硕Sentieon | 东方蜜蜂(Apis cerana)全基因组WGS分析流程
毅硕Sentieon | 电鳗(Electrophorus electricus)全基因组WGS分析流程
毅硕Sentieon | 红隼(Falco tinnunculus)全基因组WGS分析流程
毅硕Sentieon | 家猫(Felis catus)全基因组WGS分析流程
毅硕Sentieon | 尼罗罗非鱼(Oreochromis niloticus)全基因组WGS分析流程
毅硕Sentieon | 狮子(Panthera leo)全基因组WGS分析流程
毅硕Sentieon | 东北虎(panthera tigris altaica)全基因组WGS分析流程
毅硕Sentieon | 黑猩猩(pan troglodytes)全基因组WGS分析流程
毅硕Sentieon | 中国鳖(pelodiscus sinensis)全基因组WGS分析流程
毅硕Sentieon | 褐家鼠(rattus norvegicus)全基因组WGS分析流程
毅硕Sentieon | 家牛(Bos taurus)全基因WGS分析流程
毅硕Sentieon | 虎河豚(takifugu rubripes)全基因WGS分析流程
毅硕Sentieon | 人类(Homo sapiens)全基因组WGS分析流程
毅硕Sentieon文献解读
Sentieon文献解读 | Population Sequencing
Sentieon文献解读 | Agrigenomics
Sentieon | Agrigenomics-泛基因组揭示小麦结构变异与栖息地及育种的关联
Sentieon文献解读 | Genetic Disease
Sentieon文献解读 | Tumor Sequencing
Sentieon文献解读 | Benchmark and Method Study
Sentieon文献解读 | Long Read Sequencing
Sentieon文献解读 | Clinical Trial
Sentieon文献解读 | Epidemiology
Sentieon文献解读 | Gene Editing
Sentieon文献解读 | Liquid Biopsy
Sentieon | TNscope 分析流程详解
-
+
首页
Sentieon | DNAscope Hybrid长短读长混合分析流程详解及评测
# 一、前言 在基因组学研究的下半场,精确度与全面性已成为临床诊断和群体研究的核心诉求。然而,单一测序技术始终面临着“鱼与熊掌”的抉择:短读长测序(如 Illumina)虽拥有极高的碱基准确性和成本优势,但在应对结构变异(SV)、重复序列及复杂基因组区域时往往力不从心。长读长测序(如 Oxford Nanopore)凭借超长读长轻松跨越复杂区域,有效识别大型 SV,但其较高的测序成本和在同聚物(Homopolymer)区域的 Indel 错误率仍是不可忽视的屏障。 传统的混合分析方法往往只是将两种数据独立比对后进行简单的交叉验证,这不仅浪费了长读长数据中蕴含的丰富结构信息,更无法从根本上纠正复杂区域的比对偏移 。此外,长读长高昂的覆盖度要求和巨大的计算开销,让许多实验室望而却步。 Sentieon 模块化工具箱中的 sentieon-cli dnascope-hybrid 模块,通过创新的算法融合彻底打破僵局。该模块引入了创新的重比对步骤,利用长读长单倍型(Haplotype)指导短读长比对。即使在挑战性的医学相关基因(CMRG)和串联重复区域,也能实现近乎完美的变异识别。 实测数据表明,仅需 5x-10x 的低深度长读长配合标准短读长数据,其 Indel 检测准确性即可超越传统 30x 纯长读长流程,变异检测错误率降低 50% 以上。得益于 Sentieon 工业级的算法优化,该流程在 x86、arm 等多平台上运行极快,全流程分析耗时约 100 分钟,计算成本最低仅需数美元。无论是在精准医疗的临床诊断,还是在大规模的人口基因组计划中,Sentieon DNAscope Hybrid 都为您提供了一个平衡成本、速度与精度的最优解。 * * * # 二、流程总览 Sentieon® DNAscope Hybrid是一种利用单一样本的短读长和长读长测序数据结合进行种系变异识别的流程。该流程能够充分发挥两种测序技术的优势,生成的变异识别结果比单独使用短读长或长读长数据更加准确。sentieon-cli通过单条命令完整实现了DNAscope Hybrid流程。 该流程支持以下数据格式作为输入,且短读长和长读长数据均必须提供: * gzip压缩的FASTQ格式的未比对短读长数据。 * BAM或CRAM格式的已比对短读长数据。 * uBAM或uCRAM格式的未比对长读长数据。 * BAM或CRAM格式的已比对长读长数据。 流程默认生成以下输出文件: * 以VCF格式输出的小变异(单核苷酸变异和插入缺失)。 * 以VCF格式输出的结构变异。 * 以VCF格式输出的拷贝数变异。 如果输入是未比对的原始读数,流程还会输出已比对的读数,格式为BAM或CRAM。 ## 1. 硬件配置 * Sentieon® 软件包版本 202503.01 或更高。 * Python 版本 3.11 或更高。 * bcftools 版本 1.22 或更高。 * bedtools。 * MultiQC 版本 1.18 或更高,用于生成指标报告。 * samtools 版本 1.16 或更高。 * mosdepth 版本 0.2.6 或更高,用于从长读长数据中收集覆盖度指标。 ## 2. 基于已比对的短读长与长读长数据的检测胚系变异 只需运行一个命令即可从比对后的短读长序列中检测 SNV、indel、SV 和 CNV: ``` sentieon-cli dnascope-hybrid \ -r $REFERENCE \ --sr_aln $srbam \ --lr_aln $lrbam \ --rgsm RGSM \ -m $MODEL_BUNDLE \ -t $THREADS \ -g \ --skip_multiqc \ ${SAMPLEID}.vcf.gz ``` DNAscope Hybrid 流程需要以下必备参数: * `-r REFERENCE`:参考 FASTA 文件的位置。还需要一个参考 FASTA 索引文件,即“.fai”文件。 * `--sr_aln`:输入的短序列读取数据格式为 BAM 或 CRAM。可以通过在该参数后传递多个文件来提供一个或多个文件。 * `--lr_aln`:输入的长读取数据格式为 BAM 或 CRAM。可以通过在该参数后传递多个文件来提供一个或多个文件。 * `-m MODEL_BUNDLE`:模型包的位置。模型包文件位于 sentieon-models 代码库中。 * `sample.vcf.gz`:SNV 和 indel 的输出 VCF 文件的位置。该流程要求输出文件以“.vcf.gz”为后缀。 DNAscope Hybrid 流程接受以下可选参数: * `--pop_vcf POP_VCF`:包含用于 DNAModelApply 的注释信息的群体 VCF 文件的位置。某些模型包文件需要此文件。群体 VCF 文件必须与模型包匹配。 * `-d DBSNP`:用于标记已知变异的单核苷酸多态性数据库 (dbSNP) 的位置,文件格式为 VCF (`.vcf`) 或 bgzip 压缩的 VCF (`.vcf.gz`)。仅支持一个文件。提供此文件将使用变异的 dbSNP refSNP ID 编号对其进行注释。需要一个 VCF 索引文件。 * `-b DIPLOID_BED`:用于限制二倍体变异检测的参考区间,采用 BED 文件格式。提供此文件会将二倍体变异检测限制在 BED 文件内的区间范围内。 * `-t NUMBER_THREADS`:软件用于运行并行进程的计算线程数。此参数为可选参数;如果省略,则管道将使用服务器拥有的所有线程。 * `-g`:以 gVCF 格式输出变体。该工具将输出一个经过 bgzip 压缩的 gVCF 文件以及相应的索引文件。 * `-h`:打印命令行帮助并退出。 * `--dry_run`:打印管道命令,但不实际执行它们。 ## 3. 基于未比对的短读长和长读长数据中检测胚系变异 只需运行一个命令即可从未比对的短读长读序列中检测 SNV、indel、SV 和 CNV: ``` sentieon-cli dnascope-hybrid \ -r $REFERENCE \ --sr_r1_fastq HG002.novaseq.pcr-free.35x.R1.fastq.gz \ --sr_r2_fastq HG002.novaseq.pcr-free.35x.R2.fastq.gz\ --sr_readgroups "@RG\tID:HG002-1\tSM:HG002\tLB:HG002-LB-1\tPL:ILLUMINA" --lr_aln $lrbam \ --rgsm RGSM \ -m $MODEL_BUNDLE \ -t $THREADS \ -g \ --skip_multiqc \ ${SAMPLEID}.vcf.gz ``` DNAscope Hybrid 流程需要以下参数: * `--sr_r1_fastq`:输入的 R1 短读数据为 gzip 压缩的 FASTQ 格式。可以通过在该参数后传递多个文件来提供一个或多个文件。 * `--sr_r2_fastq`:输入的 R2 短读数据为 gzip 压缩的 FASTQ 格式。可以通过在该参数后添加多个文件来提供一个或多个文件。 * `--sr_readgroups`:每个 FASTQ 文件的读取组信息。该流程需要相同数量的参数传递给`--sr_r1_fastq`和 `--sr_readgroups`。例如,参数:`--sr_readgroups "@RG\tID:HG002-1\tSM:HG002\tLB:HG002-LB-1\tPL:ILLUMINA"` * `--lr_aln`:输入的长读数据,格式为 uBAM 或 uCRAM。可以通过在该参数后传递多个文件来提供一个或多个文件。 * `--lr_align_input`:指定流程对输入的长读长bam或cram进行比对,如果bam或cram是未比对的。 DNAscope Hybrid 流程接受以下可选参数: * `--sr_duplicate_marking`:重复标记设置。markdup 将标记重复的读取序列。rmdup 将删除重复的读取序列。 none 将跳过重复标记。默认设置为 markdup。 * `--lr_input_ref`:用于解码输入长读取文件的参考 fasta 文件。长读取 uCRAM 或 CRAM 输入时必需。可以与-r参数指定的 fasta 文件不同。 * `--bam_format`:对于输出对齐的文件,请使用 BAM 格式而不是 CRAM 格式。 **官方文档:** * https://support.sentieon.com/docs/sentieon\_cli/#dnascope-hybrid * * * # 三、流程输出 ## 1. DNAscope Hybrid 流程输出文件 * `sample.vcf.gz`:在-b DIPLOID_BED文件所定义的基因组区域内生成的 SNV 和 indel 变异检测结果。 * `sample.sv.vcf.gz`:来自 Sentieon® LongReadSV 工具的结构变异检测。 * `sample.cnv.vcf.gz`:来自 Sentieon® CNVscope 工具的拷贝数变异检测。 * `sample_deduped.cram`:已对齐、按坐标排序并标记重复项,从输入的 FASTQ 文件中提取短读数据。 * `sample_mm2_sorted_*.cram`:已比对和坐标排序的长读段,从输入的 uBAM、uCRAM、BAM 或 CRAM 文件中提取数据。 * `sample_metrics`:包含所分析对象的质量控制指标的目录。 **Sentieon DNAscope Hybrid 流程 github 地址:** https://github.com/Insvast/bioinformatics/tree/main/WGS-dnascope-hybird * * * # 四、实际测试运行 ## 1. Sentieon 软件下载安装 * https://ftp.insvast.com/user/Sentieon/release/sentieon-genomics-202503.03.tar.gz(适配 X86 架构 CPU 服务器,例如 Intel、 AMD、 曙光) * https://ftp.insvast.com/user/Sentieon/release/arm-sentieon-genomics-202503.03.tar.gz(适配 ARM 架构 CPU 服务器, 例如华为鲲鹏、 阿里倚天、 Ampere) * 软件下载链接需账号与密码,用户名:insvast;密码:Ins@1234。 ## 2. 参考基因组下载 ``` # 方法一: wget -c https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/references/GRCh38/GRCh38_GIABv3_no_alt_analysis_set_maskedGRC_decoys_MAP2K3_KMT2C_KCNJ18.fasta.gz # 方法二: curl -C - -0 --progress-bar https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/giab/release/references/GRCh38/GRCh38_GIABv3_no_alt_analysis_set_maskedGRC_decoys_MAP2K3_KMT2C_KCNJ18.fasta.gz # 解压 gunzip GRCh38_GIABv3_no_alt_analysis_set_maskedGRC_decoys_MAP2K3_KMT2C_KCNJ18.fasta.gz ``` ## 3. 测试数据下载 ``` wget -c https://data.nist.gov/od/ds/ark:/88434/mds2-2336/input_fastqs/HG002_35x_PacBio_14kb-15kb.fastq.gz wget -c https://data.nist.gov/od/ds/ark:/88434/mds2-2336/input_fastqs/HG002.novaseq.pcr-free.35x.R1.fastq.gz wget -c https://data.nist.gov/od/ds/ark:/88434/mds2-2336/input_fastqs/HG002.novaseq.pcr-free.35x.R2.fastq.gz wget -c https://42basepairs.com/download/gs/deepvariant/ont-case-study-testdata/HG002_R104_sup_merged.50x.bam ``` 需要注意,GIAB 早期使用的ONT数据为 R9.4.1,对 INDEL 的检出率较低,而现在一般需要使用 R10.4 版本。 ## 4. 分析运行 ``` sbatch HG002_35X_ill_10X_ONT_R104.sh sbatch HG002_35X_ill_15X_PB.sh ``` ## 5. 质控统计 * 进行分析测试的二代数据的质控信息如下表所示: |SampleID|HG002_illumina_35X| |--|--| |TotalReads|830172418| |TotalBases(Gb)|125.356035| |MappedRate(%)|99.6976| |Duplicate(%)|10.8148| |MeanDepth|31.852| |PCT_1X|98.063| |PCT_5X|97.251| |PCT_10X|96.512| |PCT_15X|94.359| |PCT_20X|90.708| |PCT_25X|83.567| |PCT_30X|65.959| |PCT_40X|17.772| |PCT_50X|1.8091| |PCT_60X|0.36279| |PCT_70X|0.23403| |PCT_80X|0.17981| |PCT_90X|0.144| |PCT_100X|0.1179| |MedianInsertSize|378| * 进行分析测试的三代数据的质控信息如下表所示: |chrom|HG002_ONT_R10.4_10X|HG002_PB_10X| |--|--|--| |length|3100432944|3100432873| |bases|29234588524|30053411531| |mean|9.43|9.69| |min|0|0| |max|4164|11578| ## 6. 分析结果一致性比较 DeepVariant 是公认的精度标杆,它开创性地将测序数据图像化并利用深度学习进行变异识别,在多项 PrecisionFDA 挑战赛中表现优异,常被视为评估新算法精度的“金标准”;而 Dragen 是一款优秀的商业软件,在行业内也广受好评。本研究将使用这两款软件与 Sentieon DNAscope Hybrid 进行性能比较。 * DeepVariant (v1.8.0)用默认设置,从仅含长读长的数据中生成变异结果。 * Dragen 的短读长准确性指标则提取自近期一项已发表研究中下载的 VCF 文件。 通过 HG002 参考样本,详细对比了 Illumina+PB 与 Illumina+ONT 数据在 SNP/Indel 检测上的准确性。下表分别列出了单核苷酸多态性(SNP)与插入缺失(Indel)的假阳性(FP)与假阴性(FN)统计指标。 * Illumina+PB 数据:各流程的变异检测准确性统计:  * Illumina+ONT 数据:各流程的变异检测准确性统计: 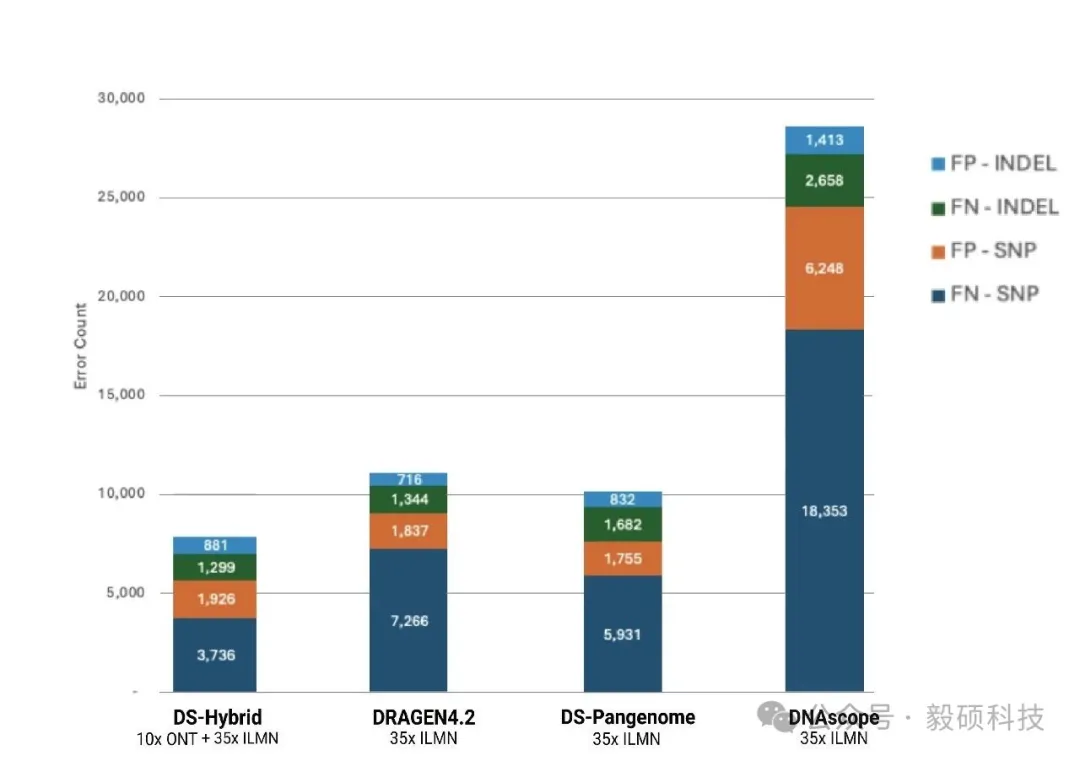 DNAscope Hybrid 的变异识别结果其准确性均高于仅使用短读长或长读长的识别结果,展现出 Sentieon DNAscope Hybrid 混测方法的稳健性与广泛适用性。未来,泛基因组信息也可集成到 DS-Hybrid 流程中,从而提供更高的准确性。 ## 7. 耗时统计 本次基准测试环境统一采用 Azure Standard HB120rs v3 实例(120个vCPU,456 GiB 内存,512GB 高级SSD),下表分别展示了在处理Illumina+PB数据和Illumina+ONT数据时,各流程的运行时间与计算成本。 其中,DNAscope Hybrid流程输出SNP/Indel/SV/CNV,DNAscope LongRead(长读长)流程输出SNP/Indel/SV,DNAscope(短读长)流程则输出SNP/Indel/CNV。 * Illumina+PB 数据:计算资源基准结果: 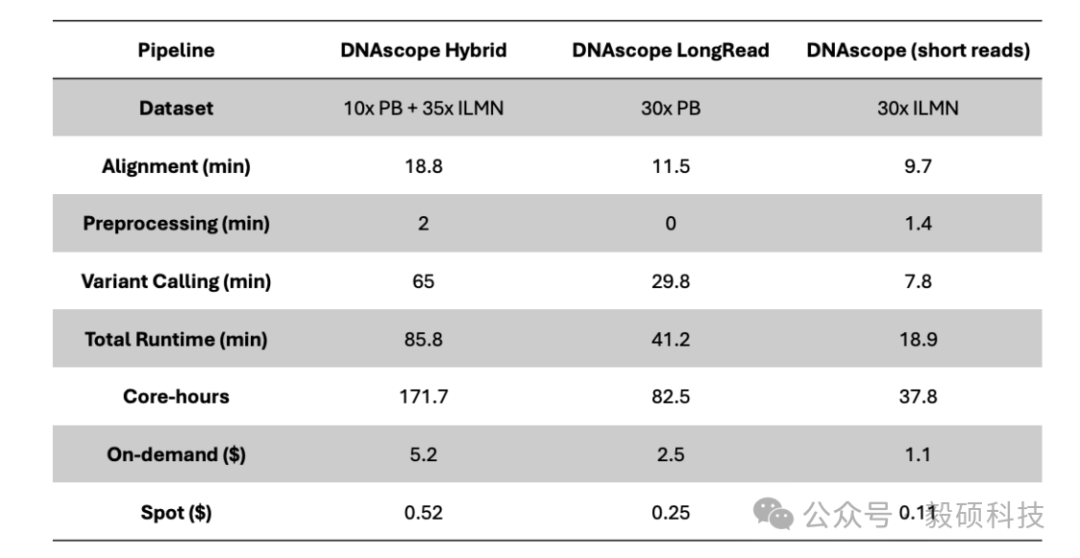 * Illumina+ONT 数据:计算资源基准结果: 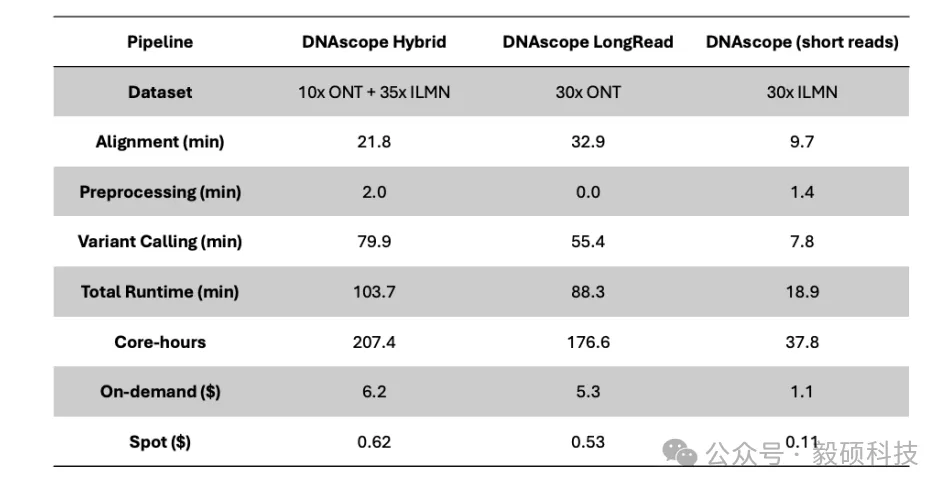 * * * # 五、总结 在 120 核 456GB 内存的测试服务器上 35X 人类 WGS 数据+10X 的 PB 数据分析仅耗时 85.8 分钟,35X 人类 WGS 数据+10X 的 ONT 数据分析仅耗时 103.7 分钟,极大缩短了分析时间,加快科研成果转化。 Sentieon 在不断的优化算法的运行效率,为科研工作者提供更快速、更经济的基因检测方案。若您刚好有需要检测的数据,不妨来申请试用 Sentieon 吧! * * * # Sentieon软件介绍 Sentieon为完整的纯软件基因变异检测二级分析方案,其分析流程完全忠于BWA、GATK、MuTect2、STAR、Minimap2、Fgbio、picard等金标准的数学模型。在匹配开源流程分析结果的前提下,大幅提升WGS、WES、Panel、UMI、ctDNA、RNA等测序数据的分析效率和检出精度,并匹配目前全部第二代、三代测序平台。 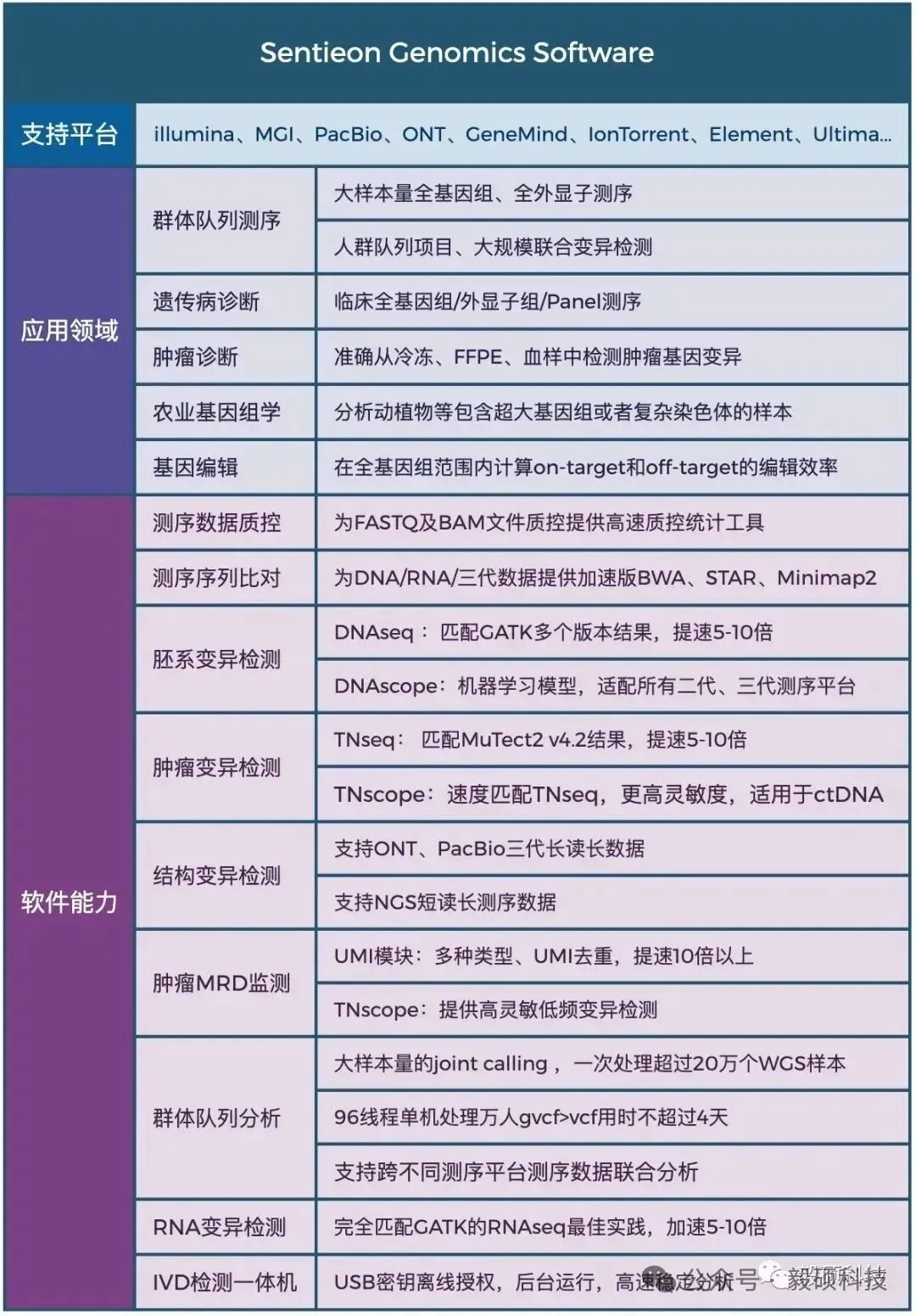 Sentieon软件团队拥有丰富的软件开发及算法优化工程经验,致力于解决生物数据分析中的速度与准确度瓶颈,**为来自于分子诊断、药物研发、临床医疗、人群队列、动植物等多个领域的合作伙伴提供高效精准的软件解决方案,共同推动基因技术的发展。** 截至2026年4月份,Sentieon已经在全球范围内为1860+用户提供服务,用户处理超过7400+PB数据量,被世界一级影响因子刊物如NEJM、Cell、Nature等广泛引用,引用次数超过1900篇。此外,Sentieon连续数年摘得了Precision FDA、Dream Challenges等多个权威评比的桂冠,在业内获得广泛认可。
chsnp
2026年4月28日 17:40
转发
收藏文档
上一篇
下一篇
手机扫码
复制链接
手机扫一扫转发分享
复制链接
Markdown文件
Word文件
PDF文档
PDF文档(打印)
分享
链接
类型
密码
更新密码
有效期