Sentieon
Sentieon 中文手册
Sentieon 中文手册(上册)
Sentieon 中文手册(下册)
Sentieon 软件应用教程
Sentieon | 应用教程: 使用DNAscope对HiFi长读长数据进行胚系变异检测分析
Sentieon | 应用教程: 利用Sentieon Python API引擎为自研算法加速
Sentieon | 应用教程: 关于读段组的建议
Sentieon | 应用教程: TNscope® 使用机器学习模型进行有匹配正常样本的体细胞变异发现
Sentieon | 应用教程: CCDG使用Sentieon®的功能等效流程
Sentieon | 应用教程: 利用共识功能去除PCR重复
Sentieon | 应用教程: 适用于PacBio HiFi和Oxford Nanopore长读长测序数据的结构变异检测
Sentieon | 应用教程: 使用 Sentieon进行大型基因组重测序分析
Sentieon | 应用教程: 体细胞SNP/Indel变异检测
Sentieon | 应用教程: DNAscope使用机器学习模型进行胚系变异调用
Sentieon | 应用教程: 唯一分子标识符(UMI)
Sentieon | 应用教程: Sentieon分布模式
Sentieon | 应用教程:使用CNVscope进行CNV检测分析
Sentieon发布核心家系(trio)基因分析最佳实践方案
Sentieon推出Segdup-caller:针对片段重复区域的专用精准变异检测工具
Sentieon软件版本更新
Sentieon | 发布V202503.01版本
Sentieon | 发布V202503.02版本
Sentieon软件快速入门指南
Sentieon 软件模块总述
Sentieon 特色流程 - DNAscope
Sentieon | DNAscope Illumina 流程
sentieon | DNAscope Complete Genomics 流程
Sentieon | DNAscope LongRead PacBio 流程
Sentieon | DNAscope Ultima Genomics 流程
Sentieon | DNAscope Element Bio 流程
Sentieon | DNAscope LongRead Nanopore 流程
Sentieon混合分析流程 - DNAscope Hybrid
Sentieon推出混合型短读长和长读长变异检测DNAscope Hybrid流程(上)
Sentieon推出混合型短读长和长读长变异检测DNAscope Hybrid流程(下)
毅硕Sentieon | 泛基因组分析流程详解
毅硕Sentieon | RNA-seq 变异检测全流程详解
毅硕Sentieon | 物种全基因组(WGS)分析流程
毅硕Sentieon | 植物全基因组(GWS)分析流程
毅硕Sentieon | 小麦(Triticum_aestivum)全基因组WGS分析流程
毅硕Sentieon | 水稻(Oryza_sativa)全基因组WGS分析流程
毅硕Sentieon | 拟南芥(Arabidopsis_thaliana)全基因组WGS分析流程
毅硕Sentieon | 马铃薯(Solanum_tuberosum)全基因组WGS分析流程
毅硕Sentieon | 巨桉(Eucalyptus grandis)全基因组WGS分析流程
毅硕Sentieon | 向日葵(Helianthus annuus)全基因组WGS分析流程
毅硕Sentieon | 野草莓(Fragaria vesca)全基因组WGS分析流程
毅硕Sentieon | 银杏(Ginkgo biloba)全基因组WGS分析流程
毅硕Sentieon | 大豆(Glycine max)全基因组WGS分析流程
毅硕Sentieon | 陆地棉(Gossypium hirsutum)全基因组WGS分析流程
毅硕Sentieon | 动物全基因组(WGS)分析流程
毅硕Sentieon | 猪(sus scrofa)全基因组WGS分析流程
毅硕Sentieon | 鸡(Gallus gallus)全基因组WGS分析流程
毅硕Sentieon | 家鼠(Mus musculus)全基因组WGS分析流程
毅硕Sentieon | 家犬(canis lupus familiaris)全基因组WGS分析流程
毅硕Sentieon | 东方蜜蜂(Apis cerana)全基因组WGS分析流程
毅硕Sentieon | 电鳗(Electrophorus electricus)全基因组WGS分析流程
毅硕Sentieon | 红隼(Falco tinnunculus)全基因组WGS分析流程
毅硕Sentieon | 家猫(Felis catus)全基因组WGS分析流程
毅硕Sentieon | 尼罗罗非鱼(Oreochromis niloticus)全基因组WGS分析流程
毅硕Sentieon文献解读
Sentieon文献解读 | Population Sequencing
Sentieon文献解读 | Agrigenomics
Sentieon | Agrigenomics-泛基因组揭示小麦结构变异与栖息地及育种的关联
Sentieon文献解读 | Genetic Disease
Sentieon文献解读 | Tumor Sequencing
Sentieon文献解读 | Benchmark and Method Study
Sentieon文献解读 | Long Read Sequencing
Sentieon文献解读 | Clinical Trial
Sentieon文献解读 | Epidemiology
Sentieon文献解读 | Gene Editing
Sentieon文献解读 | Liquid Biopsy
-
+
首页
Sentieon | 应用教程: 唯一分子标识符(UMI)
# 一、介绍 本文介绍了使用Sentieon®工具处理下一代测序数据的方法,同时利用分子条码信息(也称为唯一分子索引或UMI)。分子条码可以在测序之前在模板DNA分子的末端引入唯一标签,从而大大减少PCR重复和测序错误对变异调用过程的影响。 Sentieon®工具提供从读数据中提取UMI标签和执行基于条码的一致性生成的功能。此流程预期输入是无接头的barcode化读段数据。UMI一致性流程的输出是一个包含从barcode读数据派生出的一致性分子的BAM文件。这些一致性分子可以作为大多数变异调用软件的输入。 --- # 二、Sentieon® UMI流程 ## 1. 整体UMI流程结构 **Sentieon®提供了两个用于UMI NGS数据处理的实用工具:** - `umi extract`:从已删除接头的未比对输入读段中提取UMI标签。 - `umi consensus`:在比对输入文件上,具备条码感知的去重和一致性调用。 **Sentieon®建议使用以下典型的UMI处理流程(图1):** - 使用工具`umi extract`从未对齐的输入读段中提取UMI标签。 - 使用Sentieon®`bwa mem`将其对齐到参考基因组。 - 使用工具`umi consensus`进行UMI一致性调用。 - 使用Sentieon®`bwa mem`将一致性读段进行比对并排序。 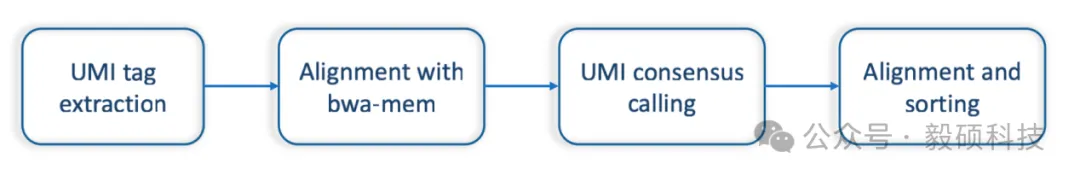 <center>图1 Sentieon® UMI处理流程</center> 以下是一个代码示例。每个组件的详细信息将在后续部分进行解释。 ``` sentieon umi extract \ 8M12S+T,+T \ sample_R1.fastq.gz \ sample_R2.fastq.gz | \ sentieon bwa mem \ -R "@RG\tID:$GROUP\tSM:$SAMPLE\tLB:$LIBRARY\tPL:$PLATFORM" \ -t $NT \ -K $BWA_K_SIZE \ -p \ -C \ $REF \ - | \ sentieon umi consensus \ -o sample_consensus.fastq.gz sentieon bwa mem \ -R "@RG\tID:$GROUP\tSM:$SAMPLE\tLB:$LIBRARY\tPL:$PLATFORM" \ -t $NT \ -K $BWA_K_SIZE \ -p \ -C \ $REF \ sample_consensus.fastq.gz | \ sentieon util sort \ -i - \ -o sample_consensus.bam \ --sam2bam --umi_post_process ``` ## 2. 确定读段结构并提取barcode序列 作为第一步,您需要从输入读段中提取barcode序列。这可以使用Sentieon®`umi extract`命令来完成,它会从读段中提取barcode序列信息,并将其添加到读段描述中。如前所述,在运行umi标签提取之前,应从输入读段中删除接头序列。这可以由其他第三方工具来完成。 `umi extract`的输出是以交错的R1和R2读段的FASTQ格式。默认情况下,提取命令的输出将发送到标准输出,除非通过选项`-o`进行其他定义。 `umi extract`命令的语法如下: ``` sentieon umi extract [options] read_structure fastq1 [fastq2] [fastq3] Options: -o Output file (default: stdout) -d Turn on duplex mode --umi_tag Logic umi tag (default 'XR') ``` `umi extract`命令的第一个参数定义了读段结构。对于成对读段,应该用逗号','分隔的两个读段结构进行指定。 读段结构由`<number><operator>`定义。数字可以是任何数字或'+',用于指示读段结尾。可能的操作符包括: - T 模板序列。 - M 分子条码序列。 - S 应忽略的一系列碱基。 使用`-d`选项可以提取双链UMI并标记其起源的链。双链UMI提取需要对两个链都指定相同的读段结构。 作为示例,以下命令演示了在成对读段上进行单端UMI提取。在这种情况下,成对读段中的第一个读段包含一个8bp的分子条码,后面是一个12bp的间隔序列,然后是模板序列。第二个读段只包含模板序列。成对读段将在输出文件中交错排列。请注意,在此示例中,输出被导向压缩为FASTQ文件的`gzip`。一般来说,我们建议将输出直接导向下一步(Sentieon®`bwa mem`)。 ``` sentieon umi extract 8M12S+T,+T \ sample_R1.fastq.gz \ sample_R2.fastq.gz | \ gzip -c \ > sample_extracted_pair.fastq.gz ``` 以下命令演示了双链UMI提取,其中两个读段均包含4bp的分子条码,后跟模板序列。 ``` sentieon umi extract \ -d \ 4M+T,4M+T \ sample_R1.fastq.gz \ sample_R2.fastq.gz | \ gzip -c \ > sample_extracted_pair.fastq.gz ``` 以下是一个使用案例,当UMI序列已经在一个单独的FASTQ文件sample_I1.fastq.gz中时。在运行这种模式下,只允许有一个额外的UMI索引读段。UMI索引读段应该不包含模板序列。这种模式不允许双链UMI提取。 ``` sentieon umi extract \ "+M,+T,+T" \ sample_I1.fastq.gz \ sample_R1.fastq.gz \ sample_R2.fastq.gz | \ gzip -c \ > sample_extracted_pair.fastq.gz ``` `umi extract`的输出包含额外的标签。默认情况下,FASTQ输出包含XR标签,该标签将用于`umi consensus`。 | 标签 | <center>意义</center> | | --- | --- | |RX | <center>提取的UMI序列碱基</center> | |XR | <center>用于在UMI一致性中分组</center> | ## 3. 参考基因组比对 可以使用`bwa mem`将fastq文件对齐到参考基因组。使用`-p`选项来指定输入文件为fastq文件,而`-C`选项将导致将fastq描述中的barcode标签附加到读段的SAM记录中。 ``` sentieon bwa mem \ -R "@RG\tID:$GROUP\tSM:$SAMPLE\tLB:$LIBRARY\tPL:$PLATFORM" \ -t $NT \ -K $BWA_K_SIZE \ -p \ -C \ $REF \ sample_extracted_pair.fastq.gz > sample_aligned.sam ``` ## 4. 共识分子的创建 流程的下一个阶段是使用Sentieon® `umi consensus`从已对齐的带barcode标记的读段中创建共识分子。 其语法如下: `umi consensus ` ``` umi consensus [-i input] [options] -o output Options for umi_consensus: -i, --input Input file (default: stdin SAM) -o, --output Output file --input_format SAM/BAM/CRAM --umi_tag Logic UMI tag (default:'XR') --copy_tags List of tags to be copied (default: XR,RX,MI,BI,BD,XZ) --read_name_prefix Read name prefix (default: 'UMI-' ) ``` 默认情况下,umi consensus将从标准输入以SAM格式读段输入。可以使用选项来覆盖默认值,指定输入文件和除SAM之外的文件格式的选项。输出是一个包含共识分子的交错的fastq文件,可以通过Sentieon®重新映射。下面是一个示例:`umi consensus` `--input` `--input_format` `umi consensus bwa mem` ``` cat sample_aligned.sam | \ sentieon umi consensus \ -o sample_consensus.fastq.gz ``` 生成的输出会生成以下额外的标签`umi consensus` | 标签 | <center>意义</center> | | --- | --- | | BI/BD | <center>插入/缺失的质量分数</center> | | MI | <center>一个唯一的标签,用于表示基于共识生成的UMI组</center> | |XZ | <center>UMI组中基于共识生成的原始读段数量。对于双工UMI,该标签将包含每个链的原始读段数量</center> | **报告没有BI/BD标签的共识读段** 默认情况下,`umi consensus`会重新校准INDEL错误率,并将这些信息存储在BI/BD标签中。可以通过从--copy_tags选项中删除BI/BD标签来关闭这个建模步骤。因此,如果不希望在报告的共识读中包含BI/BD标签,可以将这些标签从--copy_tags选项中移除。 ``` cat sample_aligned.sam | \ sentieon umi consensus \ --copy_tags XR,RX,MI,XZ \ -o sample_consensus.fastq.gz ``` ## 5. 共识读段到参考基因组的比对 可以使用Sentieon®将交错的fastq比对到参考基因组上。与之前的比对类似,选项和选项是必需的。将输出导入Sentieon®,将会生成用于变异调用的输出BAM文件。选项用于指示工具执行共识读段的必要后处理操作。`umi consensus` `bwa mem` ` -p` `-C ` `util sort` `--umi_post_process` ``` sentieon bwa mem \ -R "@RG\tID:$GROUP\tSM:$SAMPLE\tLB:$LIBRARY\tPL:$PLATFORM" \ -t $NT \ -K $BWA_K_SIZE \ -p \ -C \ $REF \ sample_consensus.fastq.gz | \ sentieon util sort \ -i - \ -o sample_consensus.bam \ --sam2bam --umi_post_process ``` ## 6. 从共识读段中进行变异调用 来自UMI共识流程的BAM文件已经准备好进行变异调用的分析。不应执行重复标记或基质质量调整的其他步骤,因为UMI共识步骤本质上是PCR重复标记和基质质量再校准的结合。 虽然可以使用任何体细胞突变调用程序与共识读段一起使用,但出于其对低频变异检测的高敏感性,我们推荐使用TNscope®。 ## 7. UMI共识读段的日志 输出的日志包含关于输入读段的统计信息,可用于质量控制。目前提供的两个信息是:`umi consensus` 组大小直方图:组大小是在一个组中支持一个共识读段的原始读段数量。该直方图显示了具有特定组大小的UMI共识读段的数量,并可用于检测问题: - 如果单体比例(组大小为1的UMI共识)过高,则很难准确建模PCR错误率并改善质量分数。 - 相反,如果每个UMI组的平均读段数过高,这可能表示文库制备中的输入DNA量过低,这将导致共识读段的覆盖范围较低。 双工统计:使用双工UMI标签,可以通过比较R1和R2读段中的UMI标签,识别来自DNA每条链的读段,并识别样本制备过程中引起的错误。日志中报告的数字表示单链或双链共识读段的数量,按以下代码进行分组:`umi consensus` - 组代码1:无互补UMI组的单链共识。 - 组代码2:有来自两个互补UMI组的双链共识。 - 组代码3:R1和R2读段的UMI序列相同。在这种情况下,依靠插入物的链确定输入读段是否来自同一链。此组进一步细分为:`umi consensus` - 组代码31:所有读段来自同一链的单链共识。 - 组代码32:来自DNA两条链的双链共识。 [**想了解更多Sentieon软件应用教程,可以点击此处进行跳转**](https://doc.insvast.com/doc/10/)
chsnp
2025年11月26日 17:32
转发
收藏文档
上一篇
下一篇
手机扫码
复制链接
手机扫一扫转发分享
复制链接
Markdown文件
Word文件
PDF文档
PDF文档(打印)
分享
链接
类型
密码
更新密码
有效期