Sentieon
Sentieon 中文手册
Sentieon 中文手册(上册)
Sentieon 中文手册(下册)
Sentieon 软件应用教程
Sentieon | 应用教程: 使用DNAscope对HiFi长读长数据进行胚系变异检测分析
Sentieon | 应用教程: 利用Sentieon Python API引擎为自研算法加速
Sentieon | 应用教程: 关于读段组的建议
Sentieon | 应用教程: TNscope® 使用机器学习模型进行有匹配正常样本的体细胞变异发现
Sentieon | 应用教程: CCDG使用Sentieon®的功能等效流程
Sentieon | 应用教程: 利用共识功能去除PCR重复
Sentieon | 应用教程: 适用于PacBio HiFi和Oxford Nanopore长读长测序数据的结构变异检测
Sentieon | 应用教程: 使用 Sentieon进行大型基因组重测序分析
Sentieon | 应用教程: 体细胞SNP/Indel变异检测
Sentieon | 应用教程: DNAscope使用机器学习模型进行胚系变异调用
Sentieon | 应用教程: 唯一分子标识符(UMI)
Sentieon | 应用教程: Sentieon分布模式
Sentieon | 应用教程:使用CNVscope进行CNV检测分析
Sentieon发布核心家系(trio)基因分析最佳实践方案
Sentieon推出Segdup-caller:针对片段重复区域的专用精准变异检测工具
Sentieon软件版本更新
Sentieon | 发布V202503.01版本
Sentieon | 发布V202503.02版本
Sentieon软件快速入门指南
Sentieon 软件模块总述
Sentieon 特色流程 - DNAscope
Sentieon | DNAscope Illumina 流程
sentieon | DNAscope Complete Genomics 流程
Sentieon | DNAscope LongRead PacBio 流程
Sentieon | DNAscope Ultima Genomics 流程
Sentieon | DNAscope Element Bio 流程
Sentieon | DNAscope LongRead Nanopore 流程
Sentieon混合分析流程 - DNAscope Hybrid
Sentieon推出混合型短读长和长读长变异检测DNAscope Hybrid流程(上)
Sentieon推出混合型短读长和长读长变异检测DNAscope Hybrid流程(下)
Sentieon | 泛基因组分析流程详解
Sentieon | 物种全基因组(WGS)分析流程
Sentieon | 植物全基因组(GWS)分析流程
毅硕Sentieon | 小麦(Triticum_aestivum)全基因组WGS分析流程
毅硕Sentieon | 水稻(Oryza_sativa)全基因组WGS分析流程
毅硕Sentieon | 拟南芥(Arabidopsis_thaliana)全基因组WGS分析流程
毅硕Sentieon | 马铃薯(Solanum_tuberosum)全基因组WGS分析流程
毅硕Sentieon | 巨桉(Eucalyptus grandis)全基因组WGS分析流程
毅硕Sentieon | 向日葵(Helianthus annuus)全基因组WGS分析流程
毅硕Sentieon | 野草莓(Fragaria vesca)全基因组WGS分析流程
毅硕Sentieon | 银杏(Ginkgo biloba)全基因组WGS分析流程
毅硕Sentieon | 大豆(Glycine max)全基因组WGS分析流程
毅硕Sentieon | 陆地棉(Gossypium hirsutum)全基因组WGS分析流程
Sentieon | 动物全基因组(WGS)分析流程
毅硕Sentieon | 猪(sus scrofa)全基因组WGS分析流程
毅硕Sentieon | 鸡(Gallus gallus)全基因组WGS分析流程
毅硕Sentieon | 家鼠(Mus musculus)全基因组WGS分析流程
毅硕Sentieon | 家犬(canis lupus familiaris)全基因组WGS分析流程
毅硕Sentieon | 东方蜜蜂(Apis cerana)全基因组WGS分析流程
毅硕Sentieon | 电鳗(Electrophorus electricus)全基因组WGS分析流程
毅硕Sentieon | 红隼(Falco tinnunculus)全基因组WGS分析流程
毅硕Sentieon | 家猫(Felis catus)全基因组WGS分析流程
毅硕Sentieon | 尼罗罗非鱼(Oreochromis niloticus)全基因组WGS分析流程
Sentieon文献解读
Sentieon文献解读 | Population Sequencing
Sentieon文献解读 | Agrigenomics
Sentieon | Agrigenomics-泛基因组揭示小麦结构变异与栖息地及育种的关联
Sentieon文献解读 | Genetic Disease
Sentieon文献解读 | Tumor Sequencing
Sentieon文献解读 | Benchmark and Method Study
Sentieon文献解读 | Long Read Sequencing
Sentieon文献解读 | Clinical Trial
Sentieon文献解读 | Epidemiology
Sentieon文献解读 | Gene Editing
Sentieon文献解读 | Liquid Biopsy
-
+
首页
Sentieon推出Segdup-caller:针对片段重复区域的专用精准变异检测工具
# 一、工具简介 **Segdup-caller 是Sentieon新开发的一款专门用于分析位于片段重复 (Segmental Duplication) 区域内的基因变异检测工具。** 该区域包括高度相似的同源基因或假基因,而这些基因会导致标准的变异检测流程在此类区域的准确性降低。 该工具的核心功能是能够利用长读长测序数据(如 PacBio HiFi 或 Oxford Nanopore)来提高变异检测的准确性,也支持使用短读长数据进行分析。 --- # 二、技术支持 **(一)支持的输入格式** - **短读长数据**:150bp双端的全基因组测序(WGS)短读长数据,如来自Illumina、Element Biosciences、MGI等平台的数据,Sentieon提供了针对不同测序平台的短读长变异检测模型。为了准确进行拷贝数检测和分型,建议最低测序深度为20X,理想深度为30X。 - **长读长数据**:支持来自PacBio HiFi或Oxford Nanopore比对后的BAM文件为额外的长读长数据输入,并使用相应的Sentieon模型进行分析来提高分析的精度。 **(二)支持的基因** Segdup-caller目前仅支持以下特定基因和片段重复区域: 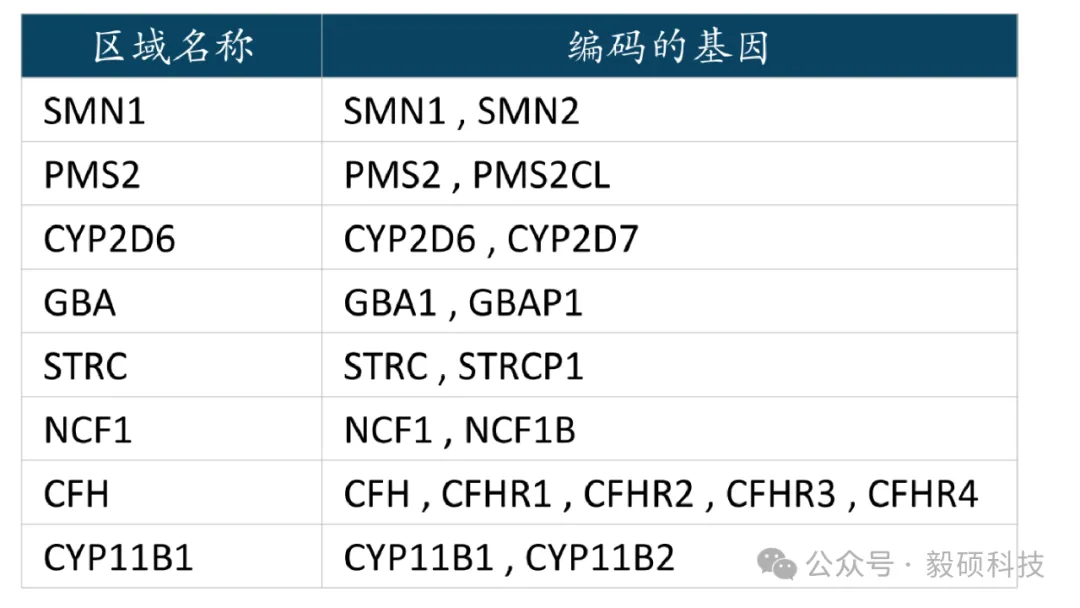 **注意**:Segdup-caller正在扩充支持的基因列表。如果您希望支持特定基因,或遇到任何问题,请提交问题反馈。 --- # 三、安装与配置 **(一)软件要求** 从Git安装软件包: ``` git clone https://github.com/Sentieon/segdup-caller.git pip install ./segdup-caller ``` **(二)运行 Segdup-caller 时需安装以下软件** - 202503或更高版本的Sentieon Genomics software - 1.16或更高版本的samtools - 1.10或更高版本的bcftools **(三)Sentieon模型** 您可以在我们的Sentieon模型包中找到模型文件:https://github.com/Sentieon/sentieon-models --- # 四、使用方法 ``` usage: segdup-caller [-h] --short SHORT [--long LONG] --sr_model SR_MODEL [--lr_model LR_MODEL] --reference REFERENCE --genes GENES [--sample_name SAMPLE_NAME] [--sr_prefix SR_PREFIX] [--lr_prefix LR_PREFIX] --outdir OUTDIR ``` 这是一个针对具有高度相似旁系同源基因的基因变异检测工具。 **选项** ``` -h,--help 显示帮助信息并退出 --short SHORT,-s SHORT 输入短读长测序数据的BAM或CRAM文件(必需) --long LONG,-l LONG 输入长读长测序数据的BAM或CRAM文件(可选) --sr_model SR_MODEL 用于短读长数据的模型包(必需) --lr_model LR_MODEL 用于长读长数据的模型包(如提供--long则必需) --reference REFERENCE,-r REFERENCE 参考基因组FASTA文件(必需) --genes GENES,-g GENES 要检测的区域列表(逗号分隔)。支持的基因 有CFH, CFHR3, CYP11B1, CYP2D6, GBA, PMS2, SMN1, STRC(必需) --sample_name SAMPLE_NAME 样本名称,默认使用短读长BAM文件中的SM标签(可选) --sr_prefix SR_PREFIX 短读长结果输出文件前缀(可选) --lr_prefix LR_PREFIX 长读长结果输出文件前缀(可选) --outdir OUTDIR,-o OUTDIR 指定输出结果的目录(必需) ``` --- ## 案例演示 以下命令展示了如何使用短读长输入BAM文件对SMN1/SMN2区域进行变异检测: ``` segdup-caller -s $short_read_bam \ -r $hg38_REF \ --sr_model $short_read_model_bundle \ -g SMN1 \ -o $outdir ``` --- # 五、输出文件 分析完成后,Segdup-caller会在指定的输出目录中产生以下文件: - VCF文件:为每个指定基因区域变异检测生成的结果文件 - YAML文件:包含额外信息(如拷贝数状态)的文件 --- # 六、总结 Sentieon推出Segdup-caller,为解决基因组中高度重复序列的分析难题提供了强有力的解决方案。segdup-caller操作简单、目标明确,特别适用于遗传病研究、药物基因组学等领域。 快来试试这款强大的新工具吧,让您的分析再无“盲区”! [**想了解更多Sentieon软件应用教程,可以点击此处进行跳转**](https://doc.insvast.com/doc/10/)
chsnp
2025年11月26日 17:35
转发
收藏文档
上一篇
下一篇
手机扫码
复制链接
手机扫一扫转发分享
复制链接
Markdown文件
Word文件
PDF文档
PDF文档(打印)
分享
链接
类型
密码
更新密码
有效期