Sentieon
Sentieon 中文手册
Sentieon 中文手册(上册)
Sentieon 中文手册(下册)
Sentieon 软件应用教程
Sentieon | 应用教程: 使用DNAscope对HiFi长读长数据进行胚系变异检测分析
Sentieon | 应用教程: 利用Sentieon Python API引擎为自研算法加速
Sentieon | 应用教程: 关于读段组的建议
Sentieon | 应用教程: TNscope® 使用机器学习模型进行有匹配正常样本的体细胞变异发现
Sentieon | 应用教程: CCDG使用Sentieon®的功能等效流程
Sentieon | 应用教程: 利用共识功能去除PCR重复
Sentieon | 应用教程: 适用于PacBio HiFi和Oxford Nanopore长读长测序数据的结构变异检测
Sentieon | 应用教程: 使用 Sentieon进行大型基因组重测序分析
Sentieon | 应用教程: 体细胞SNP/Indel变异检测
Sentieon | 应用教程: DNAscope使用机器学习模型进行胚系变异调用
Sentieon | 应用教程: 唯一分子标识符(UMI)
Sentieon | 应用教程: Sentieon分布模式
Sentieon | 应用教程:使用CNVscope进行CNV检测分析
Sentieon发布核心家系(trio)基因分析最佳实践方案
Sentieon推出Segdup-caller:针对片段重复区域的专用精准变异检测工具
Sentieon软件版本更新
Sentieon | 发布V202503.01版本
Sentieon | 发布V202503.02版本
Sentieon软件快速入门指南
Sentieon 软件模块总述
Sentieon 特色流程 - DNAscope
Sentieon | DNAscope Illumina 流程
sentieon | DNAscope Complete Genomics 流程
Sentieon | DNAscope LongRead PacBio 流程
Sentieon | DNAscope Ultima Genomics 流程
Sentieon | DNAscope Element Bio 流程
Sentieon | DNAscope LongRead Nanopore 流程
Sentieon混合分析流程 - DNAscope Hybrid
Sentieon推出混合型短读长和长读长变异检测DNAscope Hybrid流程(上)
Sentieon推出混合型短读长和长读长变异检测DNAscope Hybrid流程(下)
Sentieon | 泛基因组分析流程详解
Sentieon | 物种全基因组(WGS)分析流程
Sentieon | 植物全基因组(GWS)分析流程
Sentieon | 小麦(Triticum_aestivum)全基因组WGS分析流程
Sentieon | 水稻(Oryza_sativa)全基因组WGS分析流程
Sentieon | 拟南芥(Arabidopsis_thaliana)全基因组WGS分析流程
Sentieon | 马铃薯(Solanum_tuberosum)全基因组WGS分析流程
Sentieon | 巨桉(Eucalyptus grandis)全基因组WGS分析流程
sentieon | 向日葵(Helianthus annuus)全基因组WGS分析流程
Sentieon | 野草莓(Fragaria vesca)全基因组WGS分析流程
Sentieon | 银杏(Ginkgo biloba)全基因组WGS分析流程
Sentieon | 大豆(Glycine max)全基因组WGS分析流程
毅硕Sentieon | 陆地棉(Gossypium hirsutum)全基因组WGS分析流程
Sentieon | 动物全基因组(WGS)分析流程
Sentieon | 猪(sus scrofa)全基因组WGS分析流程
Sentieon | 鸡(Gallus gallus)全基因组WGS分析流程
Sentieon | 家鼠(Mus musculus)全基因组WGS分析流程
Sentieon | 家犬(canis lupus familiaris)全基因组WGS分析流程
sentieon | 东方蜜蜂(Apis cerana)全基因组WGS分析流程
Sentieon | 电鳗(Electrophorus electricus)全基因组WGS分析流程
Sentieon | 红隼(Falco tinnunculus)全基因组WGS分析流程
Sentieon | 家猫(Felis catus)全基因组WGS分析流程
Sentieon文献解读
Sentieon文献解读 | Population Sequencing
Sentieon文献解读 | Agrigenomics
Sentieon | Agrigenomics-泛基因组揭示小麦结构变异与栖息地及育种的关联
Sentieon文献解读 | Genetic Disease
Sentieon文献解读 | Tumor Sequencing
Sentieon文献解读 | Benchmark and Method Study
Sentieon文献解读 | Long Read Sequencing
Sentieon文献解读 | Clinical Trial
Sentieon文献解读 | Epidemiology
Sentieon文献解读 | Gene Editing
Sentieon文献解读 | Liquid Biopsy
-
+
首页
Sentieon 中文手册(下册)
[**点击此处跳转至Sentieon中文手册(上册)**](https://doc.insvast.com/doc/63/) *** ### 4.8.3 DNAscope LongRead Sentieon® DNAscope LongRead 是一套专门用于长读段测序数据的比对及胚系变异检测(包括 SNV、SV、CNV 和 Indel)的分析流程。DNAscope LongRead 流程利用长读段的长度优势,配合经过专门校准的机器学习模型,能够实现快速且准确的变异检测。sentieon-cli 通过单条命令提供了 DNAscope LongRead 流程的完整实现。该流程旨在提供卓越的准确性、高效性以及易用性。 该流程支持的输入格式包括:已比对的 BAM 或 CRAM 文件,或者未比对的 FASTQ、uBAM 或 uCRAM 文件。流程将输出 VCF(或 gVCF)格式的变异结果,以及 BAM 或 CRAM 格式的比对文件。 #### (1)环境配置 **1)必备条件** - Sentieon® 软件包:202308.01或更高版本。 - Python:3.8或更高版本。 - bcftools:1.10或更高版本(用于 SNV 和 Indel 检测流程)。 - bedtools:用于SNV和Indel检测流程。 - samtools:1.16或更高版本(用于 uBAM/uCRAM 格式的比对或对已比对数据的重新比对)。 - mosdepth:0.2.6或更高版本(用于长读段数据的覆盖度指标统计)。 - hificnv:1.0.0或更高版本(用于 CNV 检测)。 上述可执行文件均需添加至用户的PATH环境变量中,以便程序调用。 **2)参考基因组** DNAscope LongRead 将基于高质量的参考基因组序列来检测样本中的变异。除参考基因组文件外,还需提供配套的 samtools fasta 索引文件(.fai)和序列字典文件(.dict)。 我们建议对比对到不含替代重叠群的参考基因组。 #### (2) 使用说明 **1)从 FASTQ 进行比对和变异检测** 只需运行单条命令,即可完成对 FASTQ 格式的 PacBio HiFi 或 ONT 读段的比对,以及 SNV、Indel 和 SV 的检测: ``` sentieon-cli dnascope-longread [-h] \ -r REFERENCE \ --fastq INPUT_FASTQ ... \ --readgroups READGROUP ... \ -m MODEL_BUNDLE \ [-d DBSNP] \ [-b DIPLOID_BED] \ [-t NUMBER_THREADS] \ [-g] \ --tech HiFi|ONT \ [--haploid_bed HAPLOID_BED] \ [--cnv_excluded_regions CNV_EXCLUDE_BED] \ sample.vcf.gz ``` 使用 FASTQ 作为输入时,DNAscope LongRead 流程需要以下必选参数: - `-r REFERENCE`:参考基因组 FASTA 文件的路径。同时需要对应的参考序列索引 “.fai” 文件。 - `--fastq INPUT_FASTQ`:FASTQ 格式的输入样本文件。在 `--fastq` 参数后可以传入多个文件路径以同时输入一个或多个文件。 - `--readgroups READGROUP`:读段数据的读段组信息。若输入数据为FASTQ 格式,则必须提供`--readgroups`参数。该参数需要完整的读组字符串,这些字符串将通过 -R 参数传递给 minimap2 比对软件。 - 参数示例:--readgroups '@RG\tID:foo\tSM:bar'。 - `-m MODEL_BUNDLE`:模型包的路径。模型文件可在 Sentieon 官方模型仓库中找到。 - `--tech HiFi|ONT`:生成读段时所使用的测序技术。支持的参数值为 ONT或 HiFi。 - `SAMPLE_VCF`:用于存储 SNV 和 Indel 检测结果的输出 VCF 文件路径。流程要求输出文件名必须以 “.vcf.gz” 为后缀。不带后缀的文件路径将作为其他输出文件的主文件名。 Sentieon® LongRead 流程支持以下可选参数: - `-d DBSNP`:单核苷酸多态性数据库(dbSNP)的路径,用于在 VCF(.vcf 或 .vcf.gz 格式)中标记已知变异。仅支持单个文件。提供此文件将使用 dbSNP 的 refSNP ID 编号为变异添加注释。需要提供 VCF 索引文件。 - `-b DIPLOID_BED`:参考基因组上的特定区间(BED 格式),用于限制二倍体变异检测的范围。提供此文件将使二倍体变异检测仅限于 BED 文件内的区间。 - `--haploid_bed HAPLOID_BED`:参考基因组上的特定区间(BED 格式),用于限制单倍体变异检测的范围。提供此文件将在 BED 文件内的区间执行单倍体变异检测。 - `--cnv_excluded_regions`:传递给 hificnv 的 CNV 排除区域 BED 文件。更多详情请参考 hificnv (https://github.com/PacificBiosciences/HiFiCNV ) 官方文档。 - `-t NUMBER_THREADS`:软件运行并行任务时使用的线程数。此参数可选;若省略,流程将默认使用服务器的所有可用线程。 - `-g`:在输出 VCF 文件的同时,输出 gVCF 格式的变异结果。工具将输出一个经过 bgzip 压缩的 gVCF 文件及其索引文件。 - `-h`:打印命令行帮助信息并退出。 - `--dry_run`:打印流程命令,但不实际执行。 **2)从 uBAM、uCRAM、BAM 或 CRAM 进行比对和变异检测** 只需运行单条命令,即可对 uBAM、uCRAM、BAM 或 CRAM 格式的 PacBio HiFi 或 ONT 读段进行比对及 SNV、Indel 和SV的检测: ``` sentieon-cli dnascope-longread [-h] \ -r REFERENCE \ -i SAMPLE_INPUT ... \ --align \ -m MODEL_BUNDLE \ [-d DBSNP] \ [-b DIPLOID_BED] \ [-t NUMBER_THREADS] \ [-g] \ --tech HiFi|ONT \ [--haploid_bed HAPLOID_BED] \ [--cnv_excluded_regions CNV_EXCLUDE_BED] \ [--input_ref INPUT_REF] \ sample.vcf.gz ``` 使用 uBAM、uCRAM、BAM 或 CRAM 作为输入时,DNAscope LongRead流程需要以下新增参数: - `-i SAMPLE_INPUT`:uBAM 或 uCRAM 格式的输入样本文件。可以通过在 `-i` 参数后列出多个文件来提供。 - `--align`:使用 Sentieon® 优化的 minimap2 将输入读段重新比对到参考基因组。 DNAscope LongRead 流程支持以下新增可选参数: - `--input_ref INPUT_REF`:用于解码输入文件的参考基因组 FASTA 文件。在使用 uCRAM 输入时必须提供此参数。该文件可以与 `-r` 参数指定的参考基因组不同。 **3)从 BAM 或 CRAM 进行变异检测** 只需运行单条命令,即可对 BAM 或 CRAM 格式的 PacBio HiFi 或 ONT 读段进行 SNV、Indel 和 SV 的检测: ``` sentieon-cli dnascope-longread [-h] \ -r REFERENCE \ -i SAMPLE_INPUT ... \ -m MODEL_BUNDLE \ [-d DBSNP] \ [-b DIPLOID_BED] \ [-t NUMBER_THREADS] \ [-g] \ --tech HiFi|ONT \ [--haploid_bed HAPLOID_BED] \ [--cnv_excluded_regions CNV_EXCLUDE_BED] \ sample.vcf.gz ``` **注意**:若不提供 `--align`参数,流程将直接从输入的读段(即跳过比对步骤)进行变异检测。 #### (3)流程输出 在处理 FASTQ 数据,或带有`--align` 参数处理 uBAM、uCRAM、BAM、CRAM 文件时,会生成以下文件: - `sample.vcf.gz`:在 `-b DIPLOID_BED` 文件定义的基因组区域内检测到的SNV 和 Indel 变异结果。 - `sample.sv.vcf.gz`:由 Sentieon® LongReadSV 工具检测到的结构变异结果。 - `sample_mm2_sorted_fq_*.cram`:由输入 FASTQ 文件生成的经比对及坐标排序后的读段数据。 - ` sample_mm2_sorted_*.cram`:由输入的 uBAM、uCRAM、BAM 或CRAM 文件生成的经比对及坐标排序后的读段数据。 - `sample.hificnv`:HiFiCNV 输出文件的主文件名(仅在使用 HiFi 数据输入时生成)。 #### (4) 其他注意事项 **1)二倍体与单倍体变异检测** 默认流程推荐用于二倍体生物样本。对于同时包含二倍体和单倍体染色体的样本(如人类男性样本),可以使用 -b DIPLOID_BED 选项将二倍体检测限制在常染色体,并使用 --haploid_bed HAPLOID_BED 参数在单倍体染色体上执行变异检测。 本仓库的 /data 文件夹中提供了人类 hg38 参考基因组(针对男性样本)的二倍体和单倍体 BED 文件示例。 **2)脚本修改说明** 本仓库中的脚本遵循 BSD 2-Clause 开源协议。 位于 sentieon_cli/scripts 文件夹下的 Python 脚本会对流程生成的中间gVCF 和 VCF 文件进行底层处理。由于这些脚本涉及复杂的数据底层操作,不建议用户自行修改这些脚本文件。 #### (5)DNAscope LongRead PacBio HiFi分析脚本 以下是Sentieon DNAscope LongRead对PacBio HiFi数据进行全面的变异检测,包括SNPs和INDELs的分析脚本示例: ``` #!/bin/sh # # The script performs germline variant calling from PacBio HiFi reads with Sentieon DNAscope #### Argument Parsing #### die() { _ret="${2:-1}" test "${_PRINT_HELP:-no}" = yes && print_help >&2 echo "$1" >&2 exit "${_ret}" } # THE DEFAULTS INITIALIZATION - POSITIONALS # THE DEFAULTS INITIALIZATION - OPTIONALS _arg_reference_fasta= _arg_sample_input= _arg_model_bundle= _arg_dbsnp= _arg_regions= _arg_threads= _arg_gvcf="off" print_help() { printf '%s\n' "Call small variants from PacBio HiFi reads with Sentieon DNAscope" printf 'Usage: %s -r <reference> -i <sample-input> -m <model-file> [-d <dbSNP>] [-b <regions>] [-t <threads>] [-g] [-h] [--] <output-vcf>\n' "$0" printf '\t%s\n' "<output-vcf>: The output VCF file." printf '\t%s\n' "-r: The reference FASTA file. (required)" printf '\t%s\n' "-i: The aligned BAM or CRAM file. (required)" printf '\t%s\n' "-m: The model bundle file. (required)" printf '\t%s\n' "-d: dbSNP VCF file. Supplying this file will annotate variants with their dbSNP refSNP ID numbers. (no default)" printf '\t%s\n' "-b: Region BED file. Supplying this file will limit variant calling to the intervals inside the BED file. (no default)" printf '\t%s\n' "-t: Number of threads/processes to use. (number of available cores)" printf '\t%s\n' "-g: Generate a gVCF output file along with the VCF. (default generates only the VCF)" printf '\t%s\n' "-h: Print this help message" } parse_commandline() { while getopts 'r:i:m:d:b:t:gh' _key do case "$_key" in r) test "x$OPTARG" = x && die "Missing value for the argument '-$_key'." 1 _arg_reference_fasta="$OPTARG" ;; i) test "x$OPTARG" = x && die "Missing value for the argument '-$_key'." 1 _arg_sample_input="$OPTARG" ;; m) test "x$OPTARG" = x && die "Missing value for the argument '-$_key'." 1 _arg_model_bundle="$OPTARG" ;; d) test "x$OPTARG" = x && die "Missing value for the optional argument '-$_key'." 1 _arg_dbsnp="$OPTARG" ;; b) test "x$OPTARG" = x && die "Missing value for the optional argument '-$_key'." 1 _arg_regions="$OPTARG" ;; t) test "x$OPTARG" = x && die "Missing value for the optional argument '-$_key'." 1 _arg_threads="$OPTARG" ;; g) _arg_gvcf="on" ;; h) print_help exit 0 ;; *) _PRINT_HELP=yes die "FATAL ERROR: Got an unexpected option '-${_key}'" 1 ;; esac done } handle_passed_args_count() { _required_args_string="'output-vcf'" test "${_positionals_count}" -ge 1 || _PRINT_HELP=yes die "ERROR: Not enough positional arguments - we require exactly 1 (namely: $_required_args_string), but got only ${_positionals_count}." 1 test "${_positionals_count}" -le 1 || _PRINT_HELP=yes die "ERROR: There were spurious positional arguments --- we expect exactly 1 (namely: $_required_args_string), but got ${_positionals_count} (the last one was: '${_last_positional}')." 1 } assign_positional_args() { _shift_for=$1 _positional_names="_arg_output_vcf " shift "$_shift_for" for _positional_name in ${_positional_names} do test $# -gt 0 || break eval "$_positional_name=\${1}" || die "Error during argument parsing, possibly an Argbash bug." 1 shift done } parse_commandline "$@" _positionals_count=$(($# - OPTIND + 1)); _last_positional=$(eval "printf '%s' \"\$$#\""); handle_passed_args_count assign_positional_args "$OPTIND" "$@" _positionals_count=$(($# - OPTIND + 1)); _last_positional=$(eval "printf '%s' \"\$$#\""); handle_passed_args_count case "$_arg_output_vcf" in *.vcf.gz) ;; *) die "The output VCF file is expected to have a '.vcf.gz' suffix" 1 ;; esac if [ -z "$_arg_threads" ]; then _arg_threads=$(nproc) fi if [ -z "$_arg_reference_fasta" ]; then _PRINT_HELP=yes die "Error: the '-r' argument is required" 1 fi if [ -z "$_arg_sample_input" ]; then _PRINT_HELP=yes die "Error: the '-i' argument is required" 1 fi if [ -z "$_arg_model_bundle" ]; then _PRINT_HELP=yes die "Error: the '-m' argument is required" 1 fi #### Check the PATH for the required executable files #### for exec_file in bcftools bedtools sentieon; do if ! which $exec_file 1>/dev/null 2>/dev/null; then echo "Error: no '$exec_file' executable found in the PATH" exit 2 fi done #### Check executables have the required versions #### VERSION_PATTERN='^v\{0,1\}\([[:digit:]]\{1,\}\).\{0,1\}\([[:digit:]]\{1,\}\)\{0,1\}.\{0,1\}\([[:digit:]]\{1,\}\)\{0,1\}$' cmp() { num_a="$1" num_b="$2" if [ "$num_a" -lt "$num_b" ]; then return 1 elif [ "$num_a" -gt "$num_b" ]; then return 2 fi return 0 } cmp_vers() { if [ "$1" = "$2" ]; then return 0 fi a_major=$(echo $1 | sed -e 's/'"$VERSION_PATTERN"'/\1/') a_minor=$(echo $1 | sed -e 's/'"$VERSION_PATTERN"'/\2/') [ -n "$a_minor" ] || a_minor=0 a_patch=$(echo $1 | sed -e 's/'"$VERSION_PATTERN"'/\3/') [ -n "$a_patch" ] || a_patch=0 b_major=$(echo $2 | sed -e 's/'"$VERSION_PATTERN"'/\1/') b_minor=$(echo $2 | sed -e 's/'"$VERSION_PATTERN"'/\2/') [ -n "$b_minor" ] || b_minor=0 b_patch=$(echo $2 | sed -e 's/'"$VERSION_PATTERN"'/\3/') [ -n "$b_patch" ] || b_patch=0 if cmp "$a_major" "$b_major"; then if cmp "$a_minor" "$b_minor"; then cmp "$a_patch" "$b_patch" return $? else cmp "$a_minor" "$b_minor" return $? fi else cmp "$a_major" "$b_major" return $? fi } SENTIEON_MIN_VERSION=202308 BCFTOOLS_MIN_VERSION=1.10 sentieon_version_string=$(sentieon driver --version) sentieon_version_string="${sentieon_version_string##sentieon-genomics-}" bcftools_version_string=$(bcftools --version | head -n 1) bcftools_version_string="${bcftools_version_string##bcftools }" if echo "$sentieon_version_string" | grep -q "$VERSION_PATTERN"; then cmp_vers "$SENTIEON_MIN_VERSION" "$sentieon_version_string" if [ "$?" -eq "2" ]; then echo "Error: the pipeline requires sentieon version '$SENTIEON_MIN_VERSION' or later but sentieon '$sentieon_version_string' was found in the PATH" exit 2 fi else echo "Warning: unable to check sentieon version string" fi if echo "$bcftools_version_string" | grep -q "$VERSION_PATTERN"; then cmp_vers "$BCFTOOLS_MIN_VERSION" "$bcftools_version_string" if [ "$?" -eq "2" ]; then echo "Error: the pipeline requires bcftools version '$BCFTOOLS_MIN_VERSION' or later but bcftools '$bcftools_version_string' was found in the PATH" exit 2 fi else echo "Warning: unable to check bcftools version string" fi #### Check that all input files can be found and are not empty if [ ! -f "$_arg_reference_fasta" ]; then echo "Error: cannot find the input reference fasta file, '$_arg_reference_fasta'" exit 2 elif [ ! -s "$_arg_reference_fasta" ]; then echo "Error: the input reference fasta file is empty, '$_arg_reference_fasta'" exit 2 fi if [ ! -f "$_arg_sample_input" ]; then echo "Error: cannot find the input BAM file, '$_arg_sample_input'" exit 2 elif [ ! -s "$_arg_sample_input" ]; then echo "Error: the input BAM file is empty, '$_arg_sample_input'" exit 2 fi if [ ! -f "$_arg_model_bundle" ]; then echo "Error: cannot find the input model bundle file, '$_arg_model_bundle'" exit 2 elif [ ! -s "$_arg_model_bundle" ]; then echo "Error: the input model bundle file is empty, '$_arg_model_bundle'" exit 2 fi if [ -n "$_arg_dbsnp" ]; then if [ ! -f "$_arg_dbsnp" ]; then echo "Error: cannot find the optional dbSNP VCF file, '$_arg_dbsnp'" exit 2 elif [ ! -s "$_arg_dbsnp" ]; then echo "Error: the optional dbSNP VCF file is empty, '$_arg_dbsnp'" exit 2 fi fi if [ -n "$_arg_regions" ]; then if [ ! -f "$_arg_regions" ]; then echo "Error: cannot find the optional regions BED file, '$_arg_regions'" exit 2 elif [ ! -s "$_arg_regions" ]; then echo "Error: the optional regions BED file is empty, '$_arg_regions'" exit 2 fi fi #### Temporary directory #### tmp_base="/tmp" if [ -n "$SENTIEON_TMPDIR" ]; then tmp_base="$SENTIEON_TMPDIR" elif [ -n "$TMPDIR" ]; then tmp_base="$TMPDIR" fi tmp_base="$tmp_base/$$" tmp_dir="$tmp_base" while [ -e "$tmp_dir" ]; do rand_num=$(echo "" | awk 'BEGIN {srand()} {print int(rand() * 65535)}') tmp_dir="${tmp_base}"_"$rand_num" done mkdir -p "$tmp_dir" #### Output files #### OUTPUT_BASENAME=$(basename "${_arg_output_vcf%%.vcf.gz}") TMP_BASE="$tmp_dir"/"$OUTPUT_BASENAME" #### - Diploid DIPLOID_TMP_OUT="${TMP_BASE}_diploid_tmp.vcf.gz" DIPLOID_OUT="${TMP_BASE}_diploid.vcf.gz" DIPLOID_GVCF="${TMP_BASE}_diploid.g.vcf.gz" #### - Phasing PHASED_VCF="${TMP_BASE}_diploid_phased.vcf.gz" PHASED_BED="${TMP_BASE}_diploid_phased.bed" PHASED_EXT="${TMP_BASE}_diploid_phased.ext.vcf.gz" REF_BED="${TMP_BASE}_reference.bed" UNPHASED_BED="${TMP_BASE}_diploid_unphased.bed" PHASED_UNPHASED_VCF="${TMP_BASE}_diploid_phased_unphased.vcf.gz" REPEAT_MODEL="${TMP_BASE}_repeat.model" #### - Pass2 - haploid HAP_ONE_TMP="${TMP_BASE}_hap1_tmp.vcf.gz" HAP_ONE_STD_TMP="${TMP_BASE}_hap1_nohp_tmp.vcf.gz" HAP_ONE_PATCH="${TMP_BASE}_hap1_patch.vcf.gz" HAP_ONE_OUT="${TMP_BASE}_hap1.vcf.gz" HAP_TWO_TMP="${TMP_BASE}_hap2_tmp.vcf.gz" HAP_TWO_STD_TMP="${TMP_BASE}_hap2_nohp_tmp.vcf.gz" HAP_TWO_PATCH="${TMP_BASE}_hap2_patch.vcf.gz" HAP_TWO_OUT="${TMP_BASE}_hap2.vcf.gz" #### - Pass2 - diploid DIPLOID_UNPHASED_HP="${TMP_BASE}_diploid_unphased_hp.vcf.gz" DIPLOID_UNPHASED_PATCH="${TMP_BASE}_diploid_unphased_patch.vcf.gz" DIPLOID_UNPHASED="${TMP_BASE}_diploid_unphased.vcf.gz" #### - Merge generated VCF files OUTPUT_GVCF="${_arg_output_vcf%%.vcf.gz}.g.vcf.gz" #### Supporting scripts #### DIR=$(CDPATH='' cd -- "$(dirname -- "$0")" && pwd) VCF_MOD_PY="${DIR}"/vcf_mod.py GVCF_COMBINE="${DIR}"/gvcf_combine.py set -e # Un-comment this line for more log information in the Bash shell #set -exvuo pipefail #### Pipeline fuctions #### dnascope_hp() { bam="$1" model="$2" repeat_model="$3" vcf="$4" bed="$5" read_filter="${6:-}" ds_model="${7:-}" ds_vcf="${8:-}" sentieon driver -t "$_arg_threads" -r "$_arg_reference_fasta" \ --interval "$bed" -i "$bam" ${read_filter:+--read_filter $read_filter} \ ${ds_vcf:+--algo DNAscope ${_arg_dbsnp:+--dbsnp "$_arg_dbsnp"} --model "$ds_model" "$ds_vcf"} \ --algo DNAscopeHP ${_arg_dbsnp:+--dbsnp "$_arg_dbsnp"} \ --model "$model" --pcr_indel_model "$repeat_model" \ --min_repeat_count 6 "$vcf" } model_apply() { input_vcf="$1" output_vcf="$2" model="$3" sentieon driver -t "$_arg_threads" -r "$_arg_reference_fasta" \ --algo DNAModelApply --model "$model" -v "$input_vcf" "$output_vcf" } #### Pipeline #### # Pass 1 ## Call variants sentieon driver -t "$_arg_threads" -r "$_arg_reference_fasta" \ ${_arg_regions:+--interval "$_arg_regions"} -i "$_arg_sample_input" \ ${_arg_gvcf:+--algo DNAscope --model "$_arg_model_bundle"/gvcf_model --emit_mode gvcf "$DIPLOID_GVCF"} \ --algo DNAscope ${_arg_dbsnp:+--dbsnp "$_arg_dbsnp"} \ --model "$_arg_model_bundle"/diploid_model "$DIPLOID_TMP_OUT" model_apply "$DIPLOID_TMP_OUT" "$DIPLOID_OUT" "$_arg_model_bundle"/diploid_model # Phasing sentieon driver -t "$_arg_threads" -r "$_arg_reference_fasta" \ -i "$_arg_sample_input" --algo VariantPhaser -v "$DIPLOID_OUT" \ --max_depth 1000 --out_bed "$PHASED_BED" --out_ext "$PHASED_EXT" \ "$PHASED_VCF" if [ -n "$_arg_regions" ]; then bedtools subtract -a "$_arg_regions" -b "$PHASED_BED" > "$UNPHASED_BED" & else (cat "$_arg_reference_fasta".fai | awk -v OFS='\t' '{print $1,0,$2}' > "$REF_BED"; \ bedtools subtract -a "$REF_BED" -b "$PHASED_BED" > "$UNPHASED_BED") & fi ## Create the repeat model sentieon driver -t "$_arg_threads" -r "$_arg_reference_fasta" \ -i "$_arg_sample_input" --interval "$PHASED_BED" \ --read_filter PhasedReadFilter,phased_vcf="$PHASED_EXT",phase_select=tag \ --algo RepeatModel --phased --min_map_qual 1 --min_group_count 10000 \ --read_flag_mask drop=supplementary --repeat_extension 5 \ --max_repeat_unit_size 2 --min_repeat_count 6 "$REPEAT_MODEL" wait bcftools view -T "$UNPHASED_BED" "$PHASED_VCF" | \ sentieon util vcfconvert - "$PHASED_UNPHASED_VCF" & # Pass 2 - call variants on the phased haploid chromosomes ## Call variants dnascope_hp "$_arg_sample_input" "$_arg_model_bundle"/haploid_hp_model \ "$REPEAT_MODEL" "$HAP_ONE_TMP" "$PHASED_BED" \ "PhasedReadFilter,phased_vcf=$PHASED_EXT,phase_select=1" \ "$_arg_model_bundle"/haploid_model "$HAP_ONE_STD_TMP" dnascope_hp "$_arg_sample_input" "$_arg_model_bundle"/haploid_hp_model \ "$REPEAT_MODEL" "$HAP_TWO_TMP" "$PHASED_BED" \ "PhasedReadFilter,phased_vcf=$PHASED_EXT,phase_select=2" \ "$_arg_model_bundle"/haploid_model "$HAP_TWO_STD_TMP" ## Merge DNAscope and DNAscopeHP VCFs sentieon pyexec "$VCF_MOD_PY" -t "$_arg_threads" haploid_patch \ --patch1 "$HAP_ONE_PATCH" --patch2 "$HAP_TWO_PATCH" \ --hap1 "$HAP_ONE_STD_TMP" --hap2 "$HAP_TWO_STD_TMP" \ --hap1_hp "$HAP_ONE_TMP" --hap2_hp "$HAP_TWO_TMP" ## Apply the trained model to the patched VCFs model_apply "$HAP_ONE_PATCH" "$HAP_ONE_OUT" "$_arg_model_bundle"/haploid_model model_apply "$HAP_TWO_PATCH" "$HAP_TWO_OUT" "$_arg_model_bundle"/haploid_model # Pass 2 - call variant on the unphased regions dnascope_hp "$_arg_sample_input" "$_arg_model_bundle"/diploid_hp_model \ "$REPEAT_MODEL" "$DIPLOID_UNPHASED_HP" "$UNPHASED_BED" ## Merge the DNAscope and DNAscopeHP VCFs wait sentieon pyexec "$VCF_MOD_PY" -t "$_arg_threads" patch \ --vcf "$PHASED_UNPHASED_VCF" --vcf_hp "$DIPLOID_UNPHASED_HP" \ "$DIPLOID_UNPHASED_PATCH" model_apply "$DIPLOID_UNPHASED_PATCH" "$DIPLOID_UNPHASED" \ "$_arg_model_bundle"/diploid_model_unphased ## Merge the calls to create the output sentieon pyexec "$VCF_MOD_PY" -t "$_arg_threads" merge \ --hap1 "$HAP_ONE_OUT" --hap2 "$HAP_TWO_OUT" --unphased "$DIPLOID_UNPHASED" \ --phased "$PHASED_VCF" --bed "$PHASED_BED" "${_arg_output_vcf}" if [ "${_arg_gvcf}" = "on" ]; then sentieon pyexec "$GVCF_COMBINE" -t "$_arg_threads" "$DIPLOID_GVCF" "${_arg_output_vcf}" - | \ sentieon util vcfconvert - "$OUTPUT_GVCF" fi rm -r "$tmp_dir" ``` #### (6)DNAscope LongRead ONT分析脚本 以下是Sentieon DNAscope LongRead对Oxford Nanopore Technologies (ONT) 数据进行全面的变异检测,包括SNPs和INDELs的分析脚本示例: ``` #!/bin/sh # # The script performs germline variant calling from ONT long reads with Sentieon DNAscope #### Argument Parsing #### die() { _ret="${2:-1}" test "${_PRINT_HELP:-no}" = yes && print_help >&2 echo "$1" >&2 exit "${_ret}" } # THE DEFAULTS INITIALIZATION - POSITIONALS # THE DEFAULTS INITIALIZATION - OPTIONALS _arg_reference_fasta= _arg_sample_input= _arg_model_bundle= _arg_dbsnp= _arg_regions= _arg_threads= _arg_gvcf="off" print_help() { printf '%s\n' "Call small variants from ONT long reads with Sentieon DNAscope" printf 'Usage: %s -r <reference> -i <sample-input> -m <model-file> [-d <dbSNP>] [-b <regions>] [-t <threads>] [-g] [-h] [--] <output-vcf>\n' "$0" printf '\t%s\n' "<output-vcf>: The output VCF file." printf '\t%s\n' "-r: The reference FASTA file. (required)" printf '\t%s\n' "-i: The aligned BAM or CRAM file. (required)" printf '\t%s\n' "-m: The model bundle file. (required)" printf '\t%s\n' "-d: dbSNP VCF file. Supplying this file will annotate variants with their dbSNP refSNP ID numbers. (no default)" printf '\t%s\n' "-b: Region BED file. Supplying this file will limit variant calling to the intervals inside the BED file. (no default)" printf '\t%s\n' "-t: Number of threads/processes to use. (number of available cores)" printf '\t%s\n' "-g: Generate a gVCF output file along with the VCF. (default generates only the VCF)" printf '\t%s\n' "-h: Print this help message" } parse_commandline() { while getopts 'r:i:m:d:b:t:gh' _key do case "$_key" in r) test "x$OPTARG" = x && die "Missing value for the argument '-$_key'." 1 _arg_reference_fasta="$OPTARG" ;; i) test "x$OPTARG" = x && die "Missing value for the argument '-$_key'." 1 _arg_sample_input="$OPTARG" ;; m) test "x$OPTARG" = x && die "Missing value for the argument '-$_key'." 1 _arg_model_bundle="$OPTARG" ;; d) test "x$OPTARG" = x && die "Missing value for the optional argument '-$_key'." 1 _arg_dbsnp="$OPTARG" ;; b) test "x$OPTARG" = x && die "Missing value for the optional argument '-$_key'." 1 _arg_regions="$OPTARG" ;; t) test "x$OPTARG" = x && die "Missing value for the optional argument '-$_key'." 1 _arg_threads="$OPTARG" ;; g) _arg_gvcf="on" ;; h) print_help exit 0 ;; *) _PRINT_HELP=yes die "FATAL ERROR: Got an unexpected option '-${_key}'" 1 ;; esac done } handle_passed_args_count() { _required_args_string="'output-vcf'" test "${_positionals_count}" -ge 1 || _PRINT_HELP=yes die "ERROR: Not enough positional arguments - we require exactly 1 (namely: $_required_args_string), but got only ${_positionals_count}." 1 test "${_positionals_count}" -le 1 || _PRINT_HELP=yes die "ERROR: There were spurious positional arguments --- we expect exactly 1 (namely: $_required_args_string), but got ${_positionals_count} (the last one was: '${_last_positional}')." 1 } assign_positional_args() { _shift_for=$1 _positional_names="_arg_output_vcf " shift "$_shift_for" for _positional_name in ${_positional_names} do test $# -gt 0 || break eval "$_positional_name=\${1}" || die "Error during argument parsing, possibly an Argbash bug." 1 shift done } parse_commandline "$@" _positionals_count=$(($# - OPTIND + 1)); _last_positional=$(eval "printf '%s' \"\$$#\""); handle_passed_args_count assign_positional_args "$OPTIND" "$@" _positionals_count=$(($# - OPTIND + 1)); _last_positional=$(eval "printf '%s' \"\$$#\""); handle_passed_args_count case "$_arg_output_vcf" in *.vcf.gz) ;; *) die "The output VCF file is expected to have a '.vcf.gz' suffix" 1 ;; esac if [ -z "$_arg_threads" ]; then _arg_threads=$(nproc) fi if [ -z "$_arg_reference_fasta" ]; then _PRINT_HELP=yes die "Error: the '-r' argument is required" 1 fi if [ -z "$_arg_sample_input" ]; then _PRINT_HELP=yes die "Error: the '-i' argument is required" 1 fi if [ -z "$_arg_model_bundle" ]; then _PRINT_HELP=yes die "Error: the '-m' argument is required" 1 fi #### Check the PATH for the required executable files #### for exec_file in bcftools bedtools sentieon; do if ! which $exec_file 1>/dev/null 2>/dev/null; then echo "Error: no '$exec_file' executable found in the PATH" exit 2 fi done #### Check executables have the required versions #### VERSION_PATTERN='^v\{0,1\}\([[:digit:]]\{1,\}\).\{0,1\}\([[:digit:]]\{1,\}\)\{0,1\}.\{0,1\}\([[:digit:]]\{1,\}\)\{0,1\}$' cmp() { num_a="$1" num_b="$2" if [ "$num_a" -lt "$num_b" ]; then return 1 elif [ "$num_a" -gt "$num_b" ]; then return 2 fi return 0 } cmp_vers() { if [ "$1" = "$2" ]; then return 0 fi a_major=$(echo $1 | sed -e 's/'"$VERSION_PATTERN"'/\1/') a_minor=$(echo $1 | sed -e 's/'"$VERSION_PATTERN"'/\2/') [ -n "$a_minor" ] || a_minor=0 a_patch=$(echo $1 | sed -e 's/'"$VERSION_PATTERN"'/\3/') [ -n "$a_patch" ] || a_patch=0 b_major=$(echo $2 | sed -e 's/'"$VERSION_PATTERN"'/\1/') b_minor=$(echo $2 | sed -e 's/'"$VERSION_PATTERN"'/\2/') [ -n "$b_minor" ] || b_minor=0 b_patch=$(echo $2 | sed -e 's/'"$VERSION_PATTERN"'/\3/') [ -n "$b_patch" ] || b_patch=0 if cmp "$a_major" "$b_major"; then if cmp "$a_minor" "$b_minor"; then cmp "$a_patch" "$b_patch" return $? else cmp "$a_minor" "$b_minor" return $? fi else cmp "$a_major" "$b_major" return $? fi } SENTIEON_MIN_VERSION=202308 BCFTOOLS_MIN_VERSION=1.10 sentieon_version_string=$(sentieon driver --version) sentieon_version_string="${sentieon_version_string##sentieon-genomics-}" bcftools_version_string=$(bcftools --version | head -n 1) bcftools_version_string="${bcftools_version_string##bcftools }" if echo "$sentieon_version_string" | grep -q "$VERSION_PATTERN"; then cmp_vers "$SENTIEON_MIN_VERSION" "$sentieon_version_string" if [ "$?" -eq "2" ]; then echo "Error: the pipeline requires sentieon version '$SENTIEON_MIN_VERSION' or later but sentieon '$sentieon_version_string' was found in the PATH" exit 2 fi else echo "Warning: unable to check sentieon version string" fi if echo "$bcftools_version_string" | grep -q "$VERSION_PATTERN"; then cmp_vers "$BCFTOOLS_MIN_VERSION" "$bcftools_version_string" if [ "$?" -eq "2" ]; then echo "Error: the pipeline requires bcftools version '$BCFTOOLS_MIN_VERSION' or later but bcftools '$bcftools_version_string' was found in the PATH" exit 2 fi else echo "Warning: unable to check bcftools version string" fi #### Check that all input files can be found and are not empty if [ ! -f "$_arg_reference_fasta" ]; then echo "Error: cannot find the input reference fasta file, '$_arg_reference_fasta'" exit 2 elif [ ! -s "$_arg_reference_fasta" ]; then echo "Error: the input reference fasta file is empty, '$_arg_reference_fasta'" exit 2 fi if [ ! -f "$_arg_sample_input" ]; then echo "Error: cannot find the input BAM file, '$_arg_sample_input'" exit 2 elif [ ! -s "$_arg_sample_input" ]; then echo "Error: the input BAM file is empty, '$_arg_sample_input'" exit 2 fi if [ ! -f "$_arg_model_bundle" ]; then echo "Error: cannot find the input model bundle file, '$_arg_model_bundle'" exit 2 elif [ ! -s "$_arg_model_bundle" ]; then echo "Error: the input model bundle file is empty, '$_arg_model_bundle'" exit 2 fi if [ -n "$_arg_dbsnp" ]; then if [ ! -f "$_arg_dbsnp" ]; then echo "Error: cannot find the optional dbSNP VCF file, '$_arg_dbsnp'" exit 2 elif [ ! -s "$_arg_dbsnp" ]; then echo "Error: the optional dbSNP VCF file is empty, '$_arg_dbsnp'" exit 2 fi fi if [ -n "$_arg_regions" ]; then if [ ! -f "$_arg_regions" ]; then echo "Error: cannot find the optional regions BED file, '$_arg_regions'" exit 2 elif [ ! -s "$_arg_regions" ]; then echo "Error: the optional regions BED file is empty, '$_arg_regions'" exit 2 fi fi #### Temporary directory #### tmp_base="/tmp" if [ -n "$SENTIEON_TMPDIR" ]; then tmp_base="$SENTIEON_TMPDIR" elif [ -n "$TMPDIR" ]; then tmp_base="$TMPDIR" fi tmp_base="$tmp_base/$$" tmp_dir="$tmp_base" while [ -e "$tmp_dir" ]; do rand_num=$(echo "" | awk 'BEGIN {srand()} {print int(rand() * 65535)}') tmp_dir="${tmp_base}"_"$rand_num" done mkdir -p "$tmp_dir" #### Output files #### OUTPUT_BASENAME=$(basename "${_arg_output_vcf%%.vcf.gz}") TMP_BASE="$tmp_dir"/"$OUTPUT_BASENAME" #### - Diploid DIPLOID_TMP_OUT="${TMP_BASE}_diploid_tmp.vcf.gz" DIPLOID_OUT="${TMP_BASE}_diploid.vcf.gz" DIPLOID_GVCF="${TMP_BASE}_diploid.g.vcf.gz" #### - Phasing PHASED_VCF="${TMP_BASE}_diploid_phased.vcf.gz" PHASED_BED="${TMP_BASE}_diploid_phased.bed" PHASED_EXT="${TMP_BASE}_diploid_phased.ext.vcf.gz" REF_BED="${TMP_BASE}_reference.bed" UNPHASED_BED="${TMP_BASE}_diploid_unphased.bed" PHASED_PHASED_VCF="${TMP_BASE}_diploid_phased_phased.vcf.gz" PHASED_UNPHASED_VCF="${TMP_BASE}_diploid_phased_unphased.vcf.gz" REPEAT_MODEL="${TMP_BASE}_repeat.model" #### - Pass2 - haploid HAP_ONE_TMP="${TMP_BASE}_hap1_tmp.vcf.gz" HAP_ONE_PATCH="${TMP_BASE}_hap1_patch.vcf.gz" HAP_ONE_OUT="${TMP_BASE}_hap1.vcf.gz" HAP_TWO_TMP="${TMP_BASE}_hap2_tmp.vcf.gz" HAP_TWO_PATCH="${TMP_BASE}_hap2_patch.vcf.gz" HAP_TWO_OUT="${TMP_BASE}_hap2.vcf.gz" #### - Pass2 - diploid DIPLOID_UNPHASED_HP="${TMP_BASE}_diploid_unphased_hp.vcf.gz" DIPLOID_UNPHASED_PATCH="${TMP_BASE}_diploid_unphased_patch.vcf.gz" DIPLOID_UNPHASED="${TMP_BASE}_diploid_unphased.vcf.gz" #### - Merge generated VCF files OUTPUT_GVCF="${_arg_output_vcf%%.vcf.gz}.g.vcf.gz" #### Supporting scripts #### DIR=$(CDPATH='' cd -- "$(dirname -- "$0")" && pwd) VCF_MOD_PY="${DIR}"/vcf_mod.py GVCF_COMBINE="${DIR}"/gvcf_combine.py set -e # Un-comment this line for more log information in the Bash shell #set -exvuo pipefail #### Pipeline fuctions #### dnascope_hp() { bam="$1" model="$2" repeat_model="$3" vcf="$4" bed="$5" read_filter="${6:-}" sentieon driver -t "$_arg_threads" -r "$_arg_reference_fasta" \ --interval "$bed" -i "$bam" ${read_filter:+--read_filter $read_filter} \ --algo DNAscopeHP ${_arg_dbsnp:+--dbsnp "$_arg_dbsnp"} \ --model "$model" --pcr_indel_model "$repeat_model" \ --min_repeat_count 6 "$vcf" } model_apply() { input_vcf="$1" output_vcf="$2" model="$3" sentieon driver -t "$_arg_threads" -r "$_arg_reference_fasta" \ --algo DNAModelApply --model "$model" -v "$input_vcf" "$output_vcf" } #### Pipeline #### # Pass 1 ## Call variants sentieon driver -t "$_arg_threads" -r "$_arg_reference_fasta" \ ${_arg_regions:+--interval "$_arg_regions"} -i "$_arg_sample_input" \ ${_arg_gvcf:+--algo DNAscope --model "$_arg_model_bundle"/gvcf_model --emit_mode gvcf "$DIPLOID_GVCF"} \ --algo DNAscope ${_arg_dbsnp:+--dbsnp "$_arg_dbsnp"} \ --model "$_arg_model_bundle"/diploid_model "$DIPLOID_TMP_OUT" model_apply "$DIPLOID_TMP_OUT" "$DIPLOID_OUT" "$_arg_model_bundle"/diploid_model # Phasing sentieon driver -t "$_arg_threads" -r "$_arg_reference_fasta" \ -i "$_arg_sample_input" --algo VariantPhaser -v "$DIPLOID_OUT" \ --max_depth 1000 --out_bed "$PHASED_BED" --out_ext "$PHASED_EXT" \ "$PHASED_VCF" if [ -n "$_arg_regions" ]; then bedtools subtract -a "$_arg_regions" -b "$PHASED_BED" > "$UNPHASED_BED" & else (cat "$_arg_reference_fasta".fai | awk -v OFS='\t' '{print $1,0,$2}' > "$REF_BED"; \ bedtools subtract -a "$REF_BED" -b "$PHASED_BED" > "$UNPHASED_BED") & fi bcftools view -T "$PHASED_BED" "$PHASED_VCF" | \ sentieon util vcfconvert - "$PHASED_PHASED_VCF" & ## Create the repeat model sentieon driver -t "$_arg_threads" -r "$_arg_reference_fasta" \ -i "$_arg_sample_input" --interval "$PHASED_BED" \ --read_filter PhasedReadFilter,phased_vcf="$PHASED_EXT",phase_select=tag \ --algo RepeatModel --phased --min_map_qual 1 --min_group_count 10000 \ --read_flag_mask drop=supplementary --repeat_extension 5 \ --max_repeat_unit_size 2 --min_repeat_count 6 "$REPEAT_MODEL" wait bcftools view -T "$UNPHASED_BED" "$PHASED_VCF" | \ sentieon util vcfconvert - "$PHASED_UNPHASED_VCF" & # Pass 2 - call variants on the phased haploid chromosomes ## Call variants dnascope_hp "$_arg_sample_input" "$_arg_model_bundle"/haploid_hp_model \ "$REPEAT_MODEL" "$HAP_ONE_TMP" "$PHASED_BED" \ "PhasedReadFilter,phased_vcf=$PHASED_EXT,phase_select=1" dnascope_hp "$_arg_sample_input" "$_arg_model_bundle"/haploid_hp_model \ "$REPEAT_MODEL" "$HAP_TWO_TMP" "$PHASED_BED" \ "PhasedReadFilter,phased_vcf=$PHASED_EXT,phase_select=2" ## Merge DNAscope and DNAscopeHP VCFs sentieon pyexec "$VCF_MOD_PY" -t "$_arg_threads" haploid_patch \ --patch1 "$HAP_ONE_PATCH" --patch2 "$HAP_TWO_PATCH" --phased "$PHASED_PHASED_VCF" \ --hap1_hp "$HAP_ONE_TMP" --hap2_hp "$HAP_TWO_TMP" ## Apply the trained model to the patched VCFs model_apply "$HAP_ONE_PATCH" "$HAP_ONE_OUT" "$_arg_model_bundle"/haploid_model model_apply "$HAP_TWO_PATCH" "$HAP_TWO_OUT" "$_arg_model_bundle"/haploid_model # Pass 2 - call variant on the unphased regions dnascope_hp "$_arg_sample_input" "$_arg_model_bundle"/diploid_hp_model \ "$REPEAT_MODEL" "$DIPLOID_UNPHASED_HP" "$UNPHASED_BED" ## Merge the DNAscope and DNAscopeHP VCFs wait sentieon pyexec "$VCF_MOD_PY" -t "$_arg_threads" patch \ --vcf "$PHASED_UNPHASED_VCF" --vcf_hp "$DIPLOID_UNPHASED_HP" \ "$DIPLOID_UNPHASED_PATCH" model_apply "$DIPLOID_UNPHASED_PATCH" "$DIPLOID_UNPHASED" \ "$_arg_model_bundle"/diploid_model_unphased ## Merge the calls to create the output sentieon pyexec "$VCF_MOD_PY" -t "$_arg_threads" merge \ --hap1 "$HAP_ONE_OUT" --hap2 "$HAP_TWO_OUT" --unphased "$DIPLOID_UNPHASED" \ --phased "$PHASED_VCF" --bed "$PHASED_BED" "${_arg_output_vcf}" if [ "${_arg_gvcf}" = "on" ]; then sentieon pyexec "$GVCF_COMBINE" -t "$_arg_threads" "$DIPLOID_GVCF" "${_arg_output_vcf}" - | \ sentieon util vcfconvert - "$OUTPUT_GVCF" fi rm -r "$tmp_dir" ``` ### 4.8.4 DNAscopeHybrid Sentieon® DNAscope Hybrid 是一套利用同一样本的短读段与长读段组合数据进行胚系变异检测的分析流程。DNAscope Hybrid 能够结合短读段(高准确度)与长读段(长跨度、高结构解析力)技术各自的优势,在处理复杂基因组区域(如高重复序列、伪基因区)时,混合流程能显著降低假阴性并提升变异检测的完整性。sentieon-cli 通过单条命令提供了 DNAscope Hybrid 流程的完整实现。 该流程要求同时输入短读段和长读段数据,支持以下格式: - 未比对的短读段数据:Gzip 压缩格式的 FASTQ 文件。 - 已比对的短读段数据:BAM 或 CRAM 格式文件。 - 未比对的长读段数据:uBAM 或 uCRAM 格式文件。 - 已比对的长读段数据:BAM 或 CRAM 格式文件。 默认情况下,该流程将生成以下输出文件: - 小变异(SNV 和 Indel):VCF 格式。 - 结构变异(SV):VCF 格式。 - 拷贝数变异(CNV):VCF 格式。 注:若使用未比对的读段作为输入,流程还会输出比对后的 BAM 或 CRAM 文件。 #### (1)环境配置 **1)必备条件** - Sentieon® 软件包:202503.01 或更高版本。 - Python:3.8 或更高版本。 - bcftools:1.10 或更高版本。 - Bedtools。 - MultiQC:1.18 或更高版本,用于生成指标报告。 - samtools:1.16 或更高版本。 - mosdepth:0.2.6 或更高版本,用于收集长读段数据的覆盖度指标。 上述可执行文件均需添加至用户的 PATH 环境变量中,以便系统调用。 **2)参考基因组** DNAscope Hybrid 将基于 FASTA 格式的高质量参考基因组序列进行变异检测。除参考基因组外,还需提供配套的 samtools 索引文件(.fai)。短读段比对还需要 BWA 索引文件。 我们建议对比对到不含替代重叠群的参考基因组上。如果参考基因组中包含替代重叠群且流程需要执行短读段比对,请务必提供 “.alt” 文件以激活 BWA的 Alt-aware(替代序列感知)比对模式。 #### (2)使用说明 **1)从已比对的短读段与长读段数据进行胚系变异检测** 只需运行单条命令,即可从已比对的短读段、长读段中检测 SNV、Indel、SV 和CNV: ``` sentieon-cli dnascope-hybrid \ -r REFERENCE \ --sr_aln SR_ALN [SR_ALN ...] \ --lr_aln LR_ALN [LR_ALN ...] \ -m MODEL_BUNDLE \ [-b DIPLOID_BED] \ [-d DBSNP] \ [--dry_run] \ [--gvcf] \ [--sr_duplicate_marking MARKDUP] \ [-t NUMBER_THREADS] \ sample.vcf.gz ``` DNAscope Hybrid 流程需要以下必选参数: - `-r REFERENCE`:参考基因组 FASTA 文件的路径。同时需要配套的参考基因组索引文件(“.fai”)。 - `--sr_aln`:BAM 或 CRAM 格式的输入短读段数据。可以通过在 `--sr_aln` 参数后列出多个文件来提供。 - `--lr_aln`:BAM 或 CRAM 格式的输入长读段数据。可以通过在 `--lr_aln` 参数后列出多个文件来提供。 - `-m MODEL_BUNDLE`:模型包的路径。模型文件可在 Sentieon 官方模型仓库中找到。 - `sample.vcf.gz`:用于存储 SNV 和 Indel 检测结果的输出 VCF 文件路径。流程要求输出文件名必须以 “.vcf.gz” 为后缀。 DNAscope Hybrid 流程支持以下可选参数: - `-d DBSNP`:单核苷酸多态性数据库(dbSNP)的路径,用于在 VCF(.vcf或 .vcf.gz 格式)中标记已知变异,仅支持单个文件。提供此文件将使用 dbSNP 的 refSNP ID 编号为变异添加注释。需要提供 VCF 索引文件。 - `-b DIPLOID_BED`:参考基因组上的特定区间(BED 格式),用于限制二倍体变异检测的范围。提供此文件将使二倍体变异检测仅限于BED文件内的区间。 - `-t NUMBER_THREADS`:软件运行并行任务时使用的线程数。此参数为可选;若省略,流程将默认使用服务器的所有可用线程。 - `-h`:打印命令行帮助信息并退出。 - `--dry_run`:打印流程命令,但不实际执行。 **2)从未比对的短读段与长读段数据进行胚系变异检测** 只需运行单条命令,即可从未比对的短读段和长读段中检测 SNV、Indel、SV 和CNV: ``` sentieon-cli dnascope-hybrid \ -r REFERENCE \ --sr_r1_fastq SR_R1_FQ [SR_R1_FQ ...] \ --sr_r2_fastq SR_R2_FQ [SR_R2_FQ ...] \ --sr_readgroups SR_READGROUP [SR_READGROUP ...] \ --lr_aln LR_ALN [LR_ALN ...] \ --lr_align_input \ -m MODEL_BUNDLE \ [-b DIPLOID_BED] \ [--bam_format] \ [-d DBSNP] \ [--dry_run] \ [--gvcf] \ [--sr_duplicate_marking MARKDUP] \ [-t NUMBER_THREADS] \ sample.vcf.gz ``` DNAscope Hybrid 流程需要以下必选参数: - `--sr_r1_fastq`:经Gzip 压缩的输入 R1 端短读段数据。可以通过在 `--sr_r1_fastq` 参数后列出多个文件来提供。 - `--sr_r2_fastq`:经Gzip 压缩的输入 R2 端短读段数据。可以通过在 `--sr_r2_fastq` 参数后列出多个文件来提供。 - `--sr_readgroups`:每个 FASTQ 对应的读组信息。流程要求 `--sr_r1_fastq` 和 `--sr_readgroups` 的参数数量必须一致。 - 示例:--sr_readgroups"@RG\tID:HG002-1\tSM:HG002\tLB:HG002-LB-1\tPL:ILLUMINA" - `--lr_aln`:uBAM 或 uCRAM 格式的输入长读段数据。可以通过在 `--lr_aln` 后列出多个文件来提供。 - `--lr_align_input`:指示流程对比对输入的长读段。 DNAscope Hybrid 流程支持以下可选参数: - `--sr_duplicate_marking`:短读段的重复序列标记设置。`markdup` 为标记重复;`rmdup` 为移除重复;`none` 为跳过。默认设置为 `markdup`。 - ` --lr_input_ref`:用于解码输入长读段文件的参考 FASTA 文件。在使用长读段 uCRAM 或 CRAM 输入时必须提供。该文件可与 -r 参数指定的参考基因组不同。 - `--bam_format`:输出的比对文件使用 BAM 格式代替 CRAM 格式。 #### (3) 流程输出 **1)输出文件列表** DNAscope Hybrid 流程会输出以下文件: - `sample.vcf.gz`:在 `-b DIPLOID_BED` 定义的基因组区域内的 SNV 和 Indel变异结果。 - `sample.sv.vcf.gz`:来自 Sentieon® LongReadSV 工具生成的结构变异结果。 - `sample.cnv.vcf.gz`:来自 Sentieon® CNVscope 工具生成的拷贝数变异结果。 - `sample_deduped.cram`:由输入 FASTQ 生成的经比对、坐标排序及去重后的短读段数据。 - `sample_mm2_sorted_*.cram`:由输入 uBAM、uCRAM、BAM 或 CRAM 文件,经过比对和按坐标排序后的长读段数据。 - ` sample_metrics`:包含该样本所有 QC 指标的目录。 #### (4) 故障排除 如果流程检测到输入文件具有(或将要具有)不同的读组 `SM` 标签,则会触发此错误。要修复此错误,请在变异检测过程中使用 `--rgsm` 参数来统一调整输入文件的 `SM` 标签。请注意,使用此参数后,输入文件中的所有读段都将在变异检测过程中被使用。 ### 4.8.5 Sentieon Pangenome Sentieon® Pangenome 是一套利用泛基因组图谱对短读段DNA 测序数据进行比对和变异检测的分析流程。该泛基因组流程利用基于图的参考基因组表示来提高比对和变异检测的准确性,特别是在具有高序列多样性的复杂基因组区域。 该流程接受 FASTQ 格式的未比对读段或 BAM/CRAM 格式的已比对读段作为输入。流程将输出 VCF 格式的变异结果以及 BAM 或 CRAM 格式的比对文件。 #### (1)环境配置 **1)前置条件** * Sentieon® 软件:202503.02 或更高版本。 * samtools :版本 1.16 或更高,用于处理比对数据。 * vg:用于个性化泛基因组的创建及泛基因组操作。 * KMC :版本 3 或更高,用于 k-mer 计数。 * bcftools:版本 1.22 或更高,用于 VCF 相关操作 * MultiQC:用于根据流程产出的指标生成综合报告。 所有执行程序均需通过用户的 `PATH` 环境变量进行调用。 **2)参考基因组** Sentieon® Pangenome 将根据高质量的参考基因组序列进行变异检测。除参考基因组文件(FASTA)外,还需要提供 samtools fasta 索引文件 (.fai)。读段比对阶段还需要 BWA 索引文件。 我们建议使用不含替代重叠群的参考基因组。目前,该流程仅支持使用 UCSC 风格重叠群命名(如 chr1, chr2 等)的 GRCh38构建版本。 **3)泛基因组文件** 该流程需要以下泛基因组文件: * GBZ 文件:GBZ 格式的泛基因组图谱文件。 * Haplotype 文件:泛基因组单倍型信息文件。 泛基因组必须包含 GRCh38 作为参考序列。`sentieon-cli` 会检查 `.gbz` 和 `.hapl` 文件是否符合 HPRC 命名规范(即分别以 `grch38.gbz` 和 `grch38.hapl` 结尾)。该校验可以使用隐藏参数 `--skip_pangenome_name_checks` 来禁用。 **4)模型包** 需要包含用于变异检测机器学习模型的 Sentieon 模型包。模型包文件可以从 `sentieon-models` ([GitHub - Sentieon/sentieon-models: Model files for Sentieon variant callers](https://github.com/Sentieon/sentieon-models))仓库获取。 **5)群体VCF** 该流程需要一个群体 VCF 文件作为输入。该群体 VCF 必须与流程所使用的模型包相匹配。 **6)可选输入文件** 以下是实现可选功能所需的额外输入文件: * BED 文件:包含变异检测区间的 BED 文件。对于全基因组测序数据,建议使用该文件将检测限制在标准重叠群内;对于全外显子组测序数据,建议以此将检测限制在目标区域内。 * dbSNP 数据库:您希望包含在流程中的单核苷酸多态性数据库数据。数据以 VCF 格式使用,可以使用经过 bgzip 压缩并建立索引的 VCF 文件。 #### (2)使用说明 **1) 从 FASTQ 进行比对和变异检测** 运行单条命令即可从 FASTQ 文件执行比对、预处理、指标收集以及 SNV 和 Indel 检测: ``` sentieon-cli sentieon-pangenome [-h] \ -r REFERENCE \ --r1_fastq R1_FASTQ ... \ --r2_fastq R2_FASTQ ... \ --readgroups READGROUP \ --pop_vcf POP_VCF \ -m MODEL_BUNDLE \ [-b INTERVAL_FILE] \ [--bam_format\] \\ [-d DBSNP] \ [--dry_run] \ [-t NUMBER_THREADS] \ [--pcr_free] \ SAMPLE_VCF ``` 使用 FASTQ 输入时,Sentieon® 泛基因组流程接受以下必选参数: * `-r REFERENCE`:参考 FASTA 文件的路径。同时需要参考序列索引 “.fai” 文件和 BWA 索引文件。 * `--r1_fastq R1_FASTQ`:输入 R1 端 FASTQ。可多次使用。每个 R1_FASTQ 必须有对应的 R2_FASTQ 文件。所有 FASTQ 必须来自同一个样本。 * `--r2_fastq R2_FASTQ`:输入 R2 端 FASTQ。可多次使用。 * `--readgroup READGROUP`:样本的读组信息。该样本仅能提供单个读组。 * 示例参数:`--readgroup "@RG\tID:HG002-1\tSM:HG002\tLB:HG002-LB-1\tPL:ILLUMINA"`。 * `--pop_vcf POP_VCF`:群体 VCF 文件的路径。 * `-m MODEL_BUNDLE`:模型包的位置。 * `SAMPLE_VCF`:SNV 和 Indel 输出 VCF 文件的路径。要求以 `.vcf.gz` 为后缀。不带后缀的文件路径将作为其他输出文件的主文件名 。 Sentieon® 泛基因组流程接受以下可选参数: * `-b INTERVAL_FILE`:参考基因组中的区间文件,用于限制变异检测的范围,格式为 BED文件。提供此文件会将变异检测限制在 INTERVAL_FILE内的区间。我们建议使用 INTERVAL_FILE将变异检测限制在标准重叠群内。 * `--bam_format`:在比对输出文件中使用 BAM 格式代替 CRAM 格式。 * `-d DBSNP`:单核苷酸多态性数据库(dbSNP)的位置,用于在 VCF (.vcf)或 bgzip压缩的VCF (.vcf.gz)格式中标记已知变异。仅支持单个文件。提供此文件将使用 dbSNP的 refSNP ID编号为变异添加注释。需要提供 VCF 索引文件。 - `--dry_run`:打印流程命令,但不实际执行。 * `-t NUMBER_THREADS`:软件运行并行任务时使用的线程数。此参数可选;如果省略,流程将默认使用服务器的所有可用线程。 * `--pcr_free`:使用 `--pcr_indel_model NONE` 进行检测,适用于无 PCR 建库制备的文库。流程仍会执行重复序列标记(去重),以识别光学重复。 - `-h`:打印命令行帮助信息并退出。 **2) 从排序后的 BAM 或 CRAM 进行变异检测** 通过单条命令从 BAM 或 CRAM文件执行比对、预处理、指标收集以及SNV和 indel检测: ``` sentieon-cli sentieon-pangenome\[-h] \ -r REFERENCE \ -i SAMPLE_INPUT ... \ --pop_vcf POP_VCF \ -m MODEL_BUNDLE \ [-b INTERVAL_FILE] \ [--bam_format] \ [-d DBSNP] \ [--dry_run] \ [-t NUMBER_THREADS] \ SAMPLE_VCF ``` 在使用 BAM或 CRAM 输入时,泛基因组流程需要以下新增参数: * `-i SAMPLE_INPUT`:uBAM或 uCRAM格式的输入样本文件。可以通过在 `-i`参数后传递多个文件来提供一个或多个文件。 #### (3)流程输出 **1)输出文件列表** 当输出文件为 sample.vcf.gz 且启用所有功能处理全基因组测序FASTQ 数据时,将输出以下文件: * `sample.vcf.gz`:在 -b BED 文件定义的基因组区域内的 SNV 和 indel 变异检测结果。 * `sample_bwa_deduped.cram` 或 `sample_bwa_deduped.bam`:来自输入 FASTQ 文件的经过 bwa 比对、坐标排序及重复序列标记后的读段数据。 * `sample_mm2_deduped.cram` 或 `sample_mm2_deduped.bam`:来自输入 FASTQ 文件的经过泛基因组比对、坐标排序及重复序列标记后的读段数据。此文件中的 读段已比对至泛基因组并坐标回映射至参考基因组。 * `sample_metrics/`:包含被分析样本质控指标的目录。 * `--sample_metrics/sample.txt.alignment_stat.txt`:来自 Sentieon® AlignmentStat 算法的指标。 * `-- sample_metrics/sample.txt.base_distribution_by_cycle.txt`:来自 Sentieon® BaseDistributionByCycle 算法的指标。 * `-- sample_metrics/coverage*`:来自 Sentieon® CoverageMetrics 算法的覆盖度指标。 - `-- sample_metrics/sample.txt.gc_bias*`:来自 Sentieon® GCBias 算法的指标。 * `-- sample_metrics/sample.txt.insert_size.txt`:来自 Sentieon® InsertSizeMetricAlgo 算法的指标。 * `-- sample_metrics/sample.txt.mea_qual_by_cycle.txt`:来自 Sentieon® MeanQualityByCycle 算法的指标。 - `-- sample_metrics/sample.txt.qual_distribution.txt`:来自 Sentieon® QualDistribution 算法的指标。 - `-- sample_metrics/sample.txt.wgs.txt`:来自 Sentieon® WgsMetricsAlgo 算法的指标。 - `-- sample_metrics/multiqc_report.html`:由 MultiQC 汇总的收集到的质控指标。 #### (4) Sentieon® 泛基因组流程的局限性 Sentieon® 泛基因组流程目前仅支持具有 GRCh38 参考序列的 Minigraph-Cactus 泛基因组,例如由人类泛基因组参考联盟(HPRC)生成的泛基因组。 如需了解在其他泛基因组上使用该流程的信息,请联系 Sentieon® 技术支持。 ## 4.9 去重复与 UMI 处理 本章节将详细介绍如何使用 Sentieon® Genomics 工具移除 PCR 重复序列。该处理流程通过两条独立的指令协同完成,首先由系统收集读段信息,随后再执行具体的去重复操作。在此过程中,LocusCollector 模块提供的 `--consensus` 选项起到了关键作用,它负责控制系统是否输出 PCR 重复序列的共识序列。此外, 针对适用唯一分子标识符(UMI)标签的数据, 用户可以通过为LocusCollector 开启 `--umi_tag` 选项,从而激活具有条形码感知能力的去重复功能。 ### 4.9.1 非共识去重复 在“非共识去重复”模式下,系统会从一组重复读段中筛选出一个最具代表性的读段作为主要读段。 #### (1) 不含 UMI 的非共识去重复 此工作流程的运行结果与 Picard MarkDuplicates 的默认输出一致。 ``` sentieon driver \-t NUMBER\_THREADS \-i SORTED\_BAM \\ \--algo LocusCollector \--fun score\_info SCORE.gz sentieon driver \-t NUMBER\_THREADS \-i SORTED\_BAM \\ \--algo Dedup \[\--rmdup\] \--score\_info SCORE.gz \\ \--metrics DEDUP\_METRIC\_TXT DEDUPED\_BAM ``` 此外,还有一种特殊的三步走去重复流程,可用于同时标记主要读段和非主要读段。但请注意,该工作流仅适用于不含 UMI 的非共识去重复模式。 ``` sentieon driver \-t NUMBER\_THREADS \-i SORTED\_BAM \\ \--algo LocusCollector \--fun score\_info SCORE.gz sentieon driver \-t NUMBER\_THREADS \-i SORTED\_BAM \\ \--algo Dedup \--score\_info SCORE.gz \--output\_dup\_read\_name \\ \--metrics DEDUP\_METRIC\_TXT TMP\_DUP\_QNAME.gz sentieon driver \-t NUMBER\_THREADS \-i SORTED\_BAM \\ \--algo Dedup \--dup\_read\_name TMP\_DUP\_QNAME.gz \\ DEDUPED\_BAM ``` #### (2)含 UMI 的非共识去重复 该工作流除了利用读段或读段对的 5' 端位置信息外,还结合了 UMI 信息来判定 PCR 重复序列。在操作中,需通过 LocusCollector 的 `--umi_tag` 选项来指定逻辑 UMI 标签。 ``` sentieon driver \-t NUMBER\_THREADS \-i SORTED\_BAM \\ \--algo LocusCollector \--umi\_tag XR \--fun score\_info SCORE.gz sentieon driver \-t NUMBER\_THREADS \-i SORTED\_BAM \\ \--algo Dedup \[\--rmdup\] \--score\_info SCORE.gz \\ \--metrics DEDUP\_METRIC\_TXT DEDUPED\_BAM ``` ### 4.9.2 基于共识去重复 在基于共识的去重复模式下,系统会从一组重复读段中生成一条单一的共识读段。这一过程能够纠正 PCR 扩增和测序过程中引入的错误,并重新估算每个位点的碱基质量得分,以反映共识读段中碱基判定错误的最新概率。需要注意的是,在完成基于共识的去重复后,不应再进行额外的碱基质量校准。 若要开启此功能,需在 LocusCollector 中设置 `--consensus` 选项。此外,在执行后续的 Dedup 指令时,现在必须提供参考基因组的 FASTA 文件。 注意:在基于共识的去重复处理后,共识读段的配对信息可能会出现偏差。虽然Sentieon® 变异检测软件并不依赖此类配对信息,但其他生物信息学软件可能会用到。如果您的后续分析需要准确的配对信息,可以在完成去重复后使用samtools fixmate 工具对读段的配对信息进行更新。 ``` samtools collate -@ NUMBER_THREADS -Ou DEDUPED_BAM \ | samtools fixmate --reference REFERENCE -@ NUMBER_THREADS - - \ | sentieon util sort -r REFERENCE -o FIXMATE_BAM -t NUMBER_THREADS -i - ``` #### (1)不含 UMI 的基于共识去重复 在不使用 UMI 的情况下,该工作流仅依据比对后的基因组坐标对测序读段进行聚类分析。 ``` sentieon driver -t NUMBER_THREADS -i SORTED_BAM \ --algo LocusCollector --consensus --fun score_info SCORE.gz sentieon driver -t NUMBER_THREADS -r REFERENCE -i SORTED_BAM \ --algo Dedup [--rmdup] --score_info SCORE.gz \ --metrics DEDUP_METRIC_TXT DEDUPED_BAM ``` #### (2)含 UMI 的基于共识去重复 在包含 UMI 的情况下,该工作流将同时结合比对坐标及其对应的 UMI 标签对测序读段进行聚类。 ``` sentieon driver -t NUMBER_THREADS -i SORTED_BAM \ --algo LocusCollector --consensus --umi_tag XR --fun score_info SCORE.gz sentieon driver -t NUMBER_THREADS -r REFERENCE -i SORTED_BAM \ --algo Dedup [--rmdup] --score_info SCORE.gz \ --metrics DEDUP_METRIC_TXT DEDUPED_BAM ``` **1)UMI 条形码纠错** 系统会根据 UMI 条形码之间的编辑距离自动纠正其中的条形码错误。如果需要禁用此功能,可以在 LocusCollector 中使用 `--umi_ecc_dist 0` 选项。 **2)RNA 测序数据** 当处理使用 STAR 软件比对的 RNA 测序数据时,需在 LocusCollector 中设置 `--rna` 选项。 ``` sentieon driver -t NUMBER_THREADS -i SORTED_BAM \ --algo LocusCollector --rna [--consensus] [--umi_tag XR] --fun score_info SCORE.gz sentieon driver -t NUMBER_THREADS -r REFERENCE -i SORTED_BAM \ --algo Dedup [--rmdup] --score_info SCORE.gz DEDUPED_BAM ``` ### 4.9.3 附录 #### (1)确定读段结构并提取 UMI 条形码序列 作为分析的第一步,您需要从输入读段中提取条形码序列。该操作通过执行sentieon umi extract 命令完成,该命令会从读段中提取条形码序列信息并将其添加到读段描述信息中。在进行 UMI 标签提取之前,应从输入读段中移除接头序列,这一步骤可由其他第三方工具完成。 umi extract 命令的输出由 R1 和 R2 读段交错排列的 FASTQ 格式。默认情况下,提取命令的输出将发送至标准输出,除非使用 `-o` 选项另行指定输出路径。 umi extract 命令的具体语法如下: ``` sentieon umi extract [options] read_structure fastq1 [fastq2] [fastq3] Options: -o Output file (default: stdout) -d Turn on duplex mode --umi_tag Logic umi tag (default 'XR') --output_format Output format FASTQ or SAM (default 'FASTQ') ``` umi extract 命令的第一个参数用于定义读段结构。对于双端测序数据,需要指定两个由逗号“,”分隔的读段结构。 读段结构由“数字+操作符”的组合进行定义。其中数字可以是任何数字,或者使用“+”来表示读段的末尾。可选的操作符包括: - T:模板序列(即目标插入片段)。 - B:细胞条形码序列。 - M:分子条形码序列。 - S:应忽略的碱基序列。 `-d `选项用于提取双链 UMI 并标记其来源链。双链 UMI 提取要求两条链具有完全一致的读段结构。 在下方的命令中,将演示如何从双端测序读段中提取单端 UMI 的操作。在这种情况下,配对读段的 R1 端包含一个 8bp 的分子条形码,随后是一个 12bp 的间隔序列,最后是模板序列;而 R2 端仅包含模板序列。配对后的读段将以交错格式进行文件输出。请注意,在此示例中,输出结果通过管道传输给了 gzip ,以生成压缩的 FASTQ 文件。通常情况下,我们建议将输出直接通过管道传输给下一步操作(即 Sentieon® bwa mem 比对)。 ``` sentieon umi extract "8M12S+T,+T" \ sample_R1.fastq.gz \ sample_R2.fastq.gz | \ gzip -c \ > sample_extracted_pair.fastq.gz ``` 下方的命令演示了双链 UMI 的提取过程,其中双端读段均包含 4bp 的分子条形码,随后紧跟模板序列。 ``` sentieon umi extract \ -d \ "4M+T,4M+T" \ sample_R1.fastq.gz \ sample_R2.fastq.gz | \ gzip -c \ > sample_extracted_pair.fastq.gz ``` 紧接其后的代码示例则完整展示了从 UMI 提取到参考基因组比对的全过程。 ``` sentieon umi extract \ 8M12S+T,+T \ sample_R1.fastq.gz \ sample_R2.fastq.gz | \ sentieon bwa mem \ -R "@RG\tID:$GROUP\tSM:$SAMPLE\tLB:$LIBRARY\tPL:$PLATFORM" \ -t $NT \ -K $BWA_K_SIZE \ -p \ -C \ $REF \ - | \ sentieon util sort \ -i - \ -o sorted.bam \ --sam2bam ``` 此外,文档还提供了一个针对 UMI 序列已存储在独立 FASTQ 文件(如sample_I1.fastq.gz)中的应用案例。在此模式下运行分析时,仅允许额外输入一个 UMI 索引读段,且该读段中不得包含任何模板序列。需要注意的是,此模式不支持双链 UMI 的提取。 ``` sentieon umi extract \ "+M,+T,+T" \ sample_I1.fastq.gz \ sample_R1.fastq.gz \ sample_R2.fastq.gz | \ gzip -c \ > sample_extracted_pair.fastq.gz ``` 下方的指令将生成 SAM 格式的输出结果,该格式在配合使用 RNA-seq 比对软件 STAR 时非常实用。在该示例中,配对读段的 R1 端包含 16bp 的细胞条形码和 10bp 的分子条形码,而 R2 端仅包含模板序列。其最终输出结果为SAM 格式的单端读段。 ``` sentieon umi extract \ --output_format SAM \ "16B10M+S,+T" \ sample_R1.fastq.gz \ sample_R2.fastq.gz \ > sample_extracted.sam ``` umi extract 命令的输出包含额外的标签信息。在默认设置下,输出会包含一个用于记录细胞或样本条形码的 CR 标签,以及一个用于记录 UMI 序列的 XR标签,后者将供后续的 umi consensus 步骤使用。 表 4-2 由 umi extract 生成的附加标签 |标签|含义| |--|--| |RX|提取的 UMI 序列碱基| |XR|用于 umi consensus 步骤中进行分子分组的 UMI 标签| |CR|细胞条形码| ## 4.10 针对 SNPs 和 Indels 的体细胞变异检测 本文档描述了使用 TNscope® 和 TNseq® 的体细胞变异检测流程。TNscope® 使用了一种改进的变异检测算法,以获得更高的准确性和更优的运行时间;而 TNseq® 则在显著提升运行效率和并行化能力的同时,与 GATK 的 Mutect2 体细胞变异检测保持一致。 如需完整的体细胞变异检测流程,请查看手册 *4.4和4.5* 章节。 ### 4.10.1 不含唯一分子标识符(UMIs)的数据处理 #### (1)步骤 1. 比对 ``` # ****************************************** # 1a. Mapping reads with BWA-MEM, sorting for the tumor sample # ****************************************** ( sentieon bwa mem -R "@RG\tID:$tumor\tSM:$tumor\tPL:$platform" \ -t $nt -K 10000000 $fasta $tumor_fastq_1 $tumor_fastq_2 || \ echo -n 'error' ) | \ sentieon util sort -o tumor_sorted.bam -t $nt --sam2bam -i - # ****************************************** # 1b. Mapping reads with BWA-MEM, sorting for the normal sample # ****************************************** ( sentieon bwa mem -R "@RG\tID:$normal\tSM:$normal\tPL:$platform" \ -t $nt -K 10000000 $fasta $normal_fastq_1 $normal_fastq_2 || \ echo -n 'error' ) | \ sentieon util sort -o normal_sorted.bam -t $nt --sam2bam -i - ``` #### (2)步骤 2. PCR 重复序列移除(靶向扩增子测序请跳过) ``` # ****************************************** # 2a. Remove duplicate reads from the tumor sample # ****************************************** sentieon driver -t $nt -i tumor_sorted.bam \ --algo LocusCollector \ --fun score_info \ tumor_score.txt sentieon driver -t $nt -i tumor_sorted.bam \ --algo Dedup \ --score_info tumor_score.txt \ --metrics tumor_dedup_metrics.txt \ tumor_deduped.bam # ****************************************** # 2b. Remove duplicate reads from the normal sample # ****************************************** sentieon driver -t $nt -i normal_sorted.bam \ --algo LocusCollector \ --fun score_info \ normal_score.txt sentieon driver -t $nt -i normal_sorted.bam \ --algo Dedup \ --score_info normal_score.txt \ --metrics normal_dedup_metrics.txt \ normal_deduped.bam ``` #### (3)步骤 3. 碱基质量分数重校准(Panel 测序请跳过) ``` # ****************************************** # 3a. Base recalibration for the tumor sample # ****************************************** sentieon driver -r $fasta -t $nt -i tumor_deduped.bam --interval $BED \ --algo QualCal \ -k $dbsnp \ -k $known_Mills_indels \ -k $known_1000G_indels \ tumor_recal_data.table # ****************************************** # 3b. Base recalibration for the normal sample # ****************************************** sentieon driver -r $fasta -t $nt -i normal_deduped.bam --interval $BED \ --algo QualCal \ -k $dbsnp \ -k $known_Mills_indels \ -k $known_1000G_indels \ normal_recal_data.table ``` #### (4)步骤 4. 变异检测与过滤 最终变异结果集的生成分为变异检测和过滤两条命令。根据样本和文库类型的不同,应当使用不同的选项和过滤器。 以下是用于过滤的 Python 脚本 tnscope_filter.py。 ``` #!/usr/bin/env python from __future__ import print_function import argparse import heapq import itertools import sys import vcflib import operator import os from vcflib.compat import * class FilterRegistry(object): registry = [] # list of all filters @classmethod def add(cls, id, descr, params, func): params = params and params.split(',') or [] cls.registry.append((id, descr, params, func)) @classmethod def active(cls, args): return sorted([(id, descr, params, func) for id, descr, params, func in cls.registry if all( getattr(args, param, None) is not None for param in params)]) # decorator for each individual filter def Filter(id, descr, params=''): def decorator(func): FilterRegistry.add(id, descr, params, func) return func return decorator class TNscopeFilter(object): params = ( # name, defval or type, descr, metavar ('clear', 'none', 'Existing filters to clear'), ('min_qual', float, 'Minimum quality score'), ('min_depth', int, 'Minimum depth'), ('min_qd', float, 'Minimum quality by depth'), ('min_read_pos_ranksum', float, 'Minimum ReadPosRankSum'), ('min_base_qual_ranksum', float, 'Minimum BaseQRankSum'), ('max_str_length', int, 'Maximum STR length'), ('min_neighbor_base_qual', float, 'Minimum NBQPS'), ('min_map_qual_ranksum', float, 'Minimum MQRankSumPS'), ('min_depth_high_conf', float, 'Minimum ALTHC'), ('max_pv', float, 'Maximum PV'), ('max_pv2', float, 'Maximum PV2'), ('max_str_pv', float, 'Maximum PV in STR regions'), ('max_foxog', float, 'Maximum fraction of alt reads indicating OxoG'), ('max_sor', float, 'Maximum Symmetric Odds Ratio'), ('max_ecnt', int, 'Maximum number of events in this haplotype'), ('min_tumor_af', float, 'Minimum tumor allele fraction'), ('max_normal_af', float, 'Maximum normal allele fraction'), ) presets = { 'amplicon': { 'clear': 'triallelic_site', 'max_pv': 0.1, 'max_pv2': 0.1, 'max_str_pv': 0.05, 'max_foxog': 1, 'max_sor': 3, 'min_qual': 40, 'max_str_length': 10, 'max_ecnt': 10, }, 'ctdna': { 'clear': 'triallelic_site', 'min_qual': 9.225, 'min_read_pos_ranksum': -2.78, 'min_base_qual_ranksum': -5.85, 'min_depth_high_conf':3.5, 'min_qd':9.9, }, 'ctdna_umi': { 'clear': 'triallelic_site', 'min_neighbor_base_qual': 47.7, 'min_qual': 11.2, 'min_read_pos_ranksum': -2.14, 'min_base_qual_ranksum': -5.02, 'min_depth_high_conf':4.5, }, 'tissue_panel': { 'clear': 'triallelic_site', 'max_pv': 0.1, 'max_pv2': 0.1, 'max_str_pv': 0.0025, 'max_foxog': 1, 'max_sor': 3, 'min_qual': 200, 'max_str_length': 10, 'min_read_pos_ranksum': -8, 'max_normal_af': 0.01, } } @Filter('low_qual', 'Low quality score', 'min_qual') def low_qual(self, v): thr = self.args.min_qual val = v.qual return thr is not None and val is not None and val < thr @Filter('low_depth', 'Low coverage depth', 'min_depth') def low_depth(self, v): thr = self.args.min_depth val = v.samples[self.t_smid].get('AD') return thr is not None and isinstance(val, list) and sum(val) < thr @Filter('low_allele_frac', 'Low allele fraction in tumor', 'min_tumor_af') def low_allele_frac(self, v): thr = self.args.min_tumor_af val = v.samples[self.t_smid].get('AF') return thr is not None and val is not None and val < thr @Filter('alt_allele_in_normal', 'Evidence seen in the normal sample', 'max_normal_af') def high_normal_af(self, v): thr = self.args.max_normal_af val = v.samples[self.n_smid].get('AF') return self.n_smid >=0 and thr is not None and val is not None and val >= thr @Filter('low_qual_by_depth', 'Low QUAL score normalized by allele depth', 'min_qd') def low_qual_by_depth(self, v): thr = self.args.min_qd val = v.samples[self.t_smid].get('AD') if not isinstance(val, list) or v.qual is None or thr is None: return False return v.qual < thr * sum(val[1:]) @Filter('read_pos_bias', 'Alt vs. Ref read position bias', 'min_read_pos_ranksum') def read_pos_bias(self, v): thr = self.args.min_read_pos_ranksum val = v.samples[self.t_smid].get('ReadPosRankSumPS') return thr is not None and val is not None and val < thr @Filter('base_qual_bias', 'Alt Vs. Ref base quality bias', 'min_base_qual_ranksum') def base_qual_bias(self, v): thr = self.args.min_base_qual_ranksum val = v.samples[self.t_smid].get('BaseQRankSumPS') return thr is not None and val is not None and val < thr @Filter('neighbor_base_qual', 'Low mean neighboring base quality', 'min_neighbor_base_qual') def neighbor_base_qual(self, v): thr = self.args.min_neighbor_base_qual val = v.samples[self.t_smid].get('NBQPS') return thr is not None and val is not None and val < thr @Filter('map_qual_bias', 'Alt Vs. Ref mapping qualities bias', 'min_map_qual_ranksum') def z_score_wilcoxon(self, v): thr = self.args.min_z_score_wilcoxon val = v.samples[self.t_smid].get('MQRankSumPS') return thr is not None and val is not None and val < thr @Filter('depth_high_conf', 'Depth high conf', 'min_depth_high_conf') def depth_high_conf(self, v): thr = self.args.min_depth_high_conf val = v.samples[self.t_smid].get('ALTHC') return thr is not None and val is not None and val < thr @Filter('short_tandem_repeat', 'Short tandem repeat', 'max_str_length') def short_tandem_repeat(self, v): thr = self.args.max_str_length val = v.info.get('RPA') return thr is not None and isinstance(val, list) and val[0] >= thr @Filter('insignificant', 'Insignificant call', 'max_pv,max_pv2') def insignificant(self, v): thr1 = self.args.max_pv thr2 = self.args.max_pv2 val1 = v.info.get('PV') val2 = v.info.get('PV2') return thr1 is not None and thr2 is not None and val1 is not None and val1 > thr1 and val2 is not None and val2 > thr2 @Filter('insignificant_str', 'Insignificant call in STR regions', 'max_str_pv') def insignificant_str(self, v): thr = self.args.max_str_pv val = v.info.get('PV') return v.info.get('STR') is not None and thr is not None and val is not None and val > thr @Filter('orientation_bias', 'Orientation bias', 'max_foxog') def orientation_bias(self, v): thr = self.args.max_foxog val = v.samples[self.t_smid].get('FOXOG') return thr is not None and val is not None and val >= thr @Filter('strand_bias', 'Strand bias', 'max_sor') def strand_bias(self, v): thr = self.args.max_sor val = v.info.get('SOR') return thr is not None and val is not None and val > thr @Filter('noisy_region', 'Variant in noisy region', 'max_ecnt') def noisy_region(self, v): thr = self.args.max_ecnt val = v.info.get('ECNT') return thr is not None and val is not None and val > thr @classmethod def add_arguments(cls, parser, preset=None): vals = {} if preset is not None: vals = cls.presets.get(preset, vals) if cls.presets: v = cls.presets.keys() h = 'Parameter preset (choices: '+','.join(v)+')' parser.add_argument('-x', '--preset', choices=v, type=str, help=h) for k,v,h in cls.params: v = vals.get(k, v) if isinstance(v, type): h = argparse.SUPPRESS if vals else h parser.add_argument('--'+k, type=v, help=h) elif v is not None: h += ' (default: %(default)s)' parser.add_argument('--'+k, default=v, type=type(v), help=h) @staticmethod def grouper(*vcfs): q = [] for k, vcf in enumerate(vcfs): i = iter(vcf) v = next(i, None) if v: heapq.heappush(q, (v.pos, v.end, k, v, i)) while q: grp = [[] for _ in vcfs] pos, end, k, v, i = heapq.heappop(q) grp[k].append(v) v = next(i, None) if v: heapq.heappush(q, (v.pos, v.end, k, v, i)) while q and q[0][0] == pos: pos, end, k, v, i = heapq.heappop(q) grp[k].append(v) v = next(i, None) if v: heapq.heappush(q, (v.pos, v.end, k, v, i)) yield (pos, grp) def __init__(self, args, t_smid, n_smid): self.args = args self.t_smid = t_smid self.n_smid = n_smid self.filters = FilterRegistry.active(args) if args.clear == 'all': self.clear = None elif args.clear == 'none' or args.clear == '': self.clear = set() self.clear.add('PASS') else: self.clear = set(args.clear.split(',')) self.clear.add('PASS') def apply(self, invcf, outvcf): self.copy_header(invcf, outvcf) for chrom in invcf.contigs.keys(): for pos, grp in self.grouper(invcf.range(chrom)): if len(grp[0]) > 1: grp[0].sort(key=operator.attrgetter('qual'), reverse=True) for v in grp[0]: outvcf.emit(self.apply_filters(v)) def apply_filters(self, v): filters = set(id for id, _, _, func in self.filters if func(self, v)) if self.clear is not None: filters = set(v.filter) - self.clear | filters v.filter = sorted(filters) flds = v.line.split('\t') flds[6] = v.filter and ';'.join(v.filter) or 'PASS' v.line = '\t'.join(flds) return v def copy_header(self, invcf, outvcf): fmt = '##FILTER=<ID=%s,Description="%s">' idg = itertools.groupby(self.filters, key=operator.itemgetter(0)) toupdate = [fmt % next(g)[:2] for id, g in idg] if self.clear is None: clear = set(invcf.filters.keys()) else: clear = set(invcf.filters.keys()) & self.clear clear.discard('PASS') toremove = [fmt % (id, '') for id in clear] outvcf.copy_header(invcf, toupdate, toremove) outvcf.emit_header() def main(args): if not os.path.exists(args.vcf): print('Error: input file %s does not exist' % args.vcf, file=sys.stderr) return -1 invcf = vcflib.VCF(args.vcf, 'r') try: t_smid = invcf.samples.index(args.tumor_sample) except ValueError: print('Error: tumor sample "%s" not in input file %s' % (args.tumor_sample, args.vcf), file=sys.stderr) return -1 try: if args.normal_sample: n_smid = invcf.samples.index(args.normal_sample) else: n_smid = -1 except ValueError: print('Error: normal sample "%s" not in input file %s' % (args.normal_sample, args.vcf), file=sys.stderr) return -1 outvcf = vcflib.VCF(args.output, 'w') filter = TNscopeFilter(args, t_smid, n_smid) filter.apply(invcf, outvcf) outvcf.close() invcf.close() return 0 class MixedHelpFormatter(argparse.HelpFormatter): def _format_usage(self, usage, actions, groups, prefix): if prefix is None: prefix = 'usage: sentieon pyexec ' return argparse.HelpFormatter._format_usage( self, usage, actions, groups, prefix) def _metavar_formatter(self, action, default): if action.metavar is None and action.type is not None: action.metavar = action.type.__name__.upper() return argparse.HelpFormatter._metavar_formatter( self, action, default) if __name__ == '__main__': parser = argparse.ArgumentParser(add_help=False) parser.add_argument('-x', '--preset', const='?', nargs='?') args, argv = parser.parse_known_args() parser = argparse.ArgumentParser(formatter_class=MixedHelpFormatter) parser.add_argument('output', help='Output vcf file name') parser.add_argument('-v', '--vcf', required=True, help='Input vcf file name') parser.add_argument('--tumor_sample', required=True, help='Tumor sample name', type=str) parser.add_argument('--normal_sample', help='Normal sample name', type=str) TNscopeFilter.add_arguments(parser, args.preset) if args.preset == '?': argv = ['-x'] + argv elif args.preset is not None: argv = ['-x', args.preset] + argv sys.exit(main(parser.parse_args(argv))) # vim: ts=4 sw=4 expandtab ``` TNseq 针对全基因组(WGS)及全外显子组(WES)的变异检测具体脚本可查阅手册第 *4.4 TNseq®* 章节;TNscope 针对特定样本类型的变异检测优化逻辑与脚本方案,可查阅*4.5 TNscope®* 章节; ## 4.11 Joint calling ### 4.11.1 Joint calling分析脚本 以下是一个完整的DNA测序数据特别是针对多个样本的联合变异检测(joint calling)的分析脚本示例: ``` #!/bin/sh # Copyright (c) 2016-2020 Sentieon Inc. All rights reserved # ******************************************* # Script to perform joint calling on 3 samples # with fastq files named sample<i>_1.fastq.gz # and sample<i>_2.fastq.gz # ******************************************* set -eu # Update with the fullpath location of your sample fastq SM_LIST="sample1 sample2 sample3" # list of sample names PL="ILLUMINA" #or other sequencing platform FASTQ_FOLDER="/home/pipeline/samples" FASTQ_1_SUFFIX="1.fastq.gz" FASTQ_2_SUFFIX="2.fastq.gz" #If using Illumina paired data # Update with the location of the reference data files FASTA_DIR="/home/regression/references/b37/" FASTA="$FASTA_DIR/human_g1k_v37_decoy.fasta" KNOWN_DBSNP="$FASTA_DIR/dbsnp_138.b37.vcf.gz" KNOWN_INDELS="$FASTA_DIR/1000G_phase1.indels.b37.vcf.gz" KNOWN_MILLS="$FASTA_DIR/Mills_and_1000G_gold_standard.indels.b37.vcf.gz" # Update with the location of the Sentieon software package and license file SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-|release_version| export SENTIEON_LICENSE=/home/Licenses/Sentieon.lic #or using licsrvr: c1n11.sentieon.com:5443 # Other settings NT=$(nproc) #number of threads to use in computation, set to number of cores in the server START_DIR="$PWD/test/DNAseq_joint" #Determine where the output files will be stored # You do not need to modify any of the lines below unless you want to tweak the pipeline # ************************************************************************************************************************************************************************ # ****************************************** # 0. Setup # ****************************************** WORKDIR="$START_DIR/joint_call" mkdir -p $WORKDIR LOGFILE=$WORKDIR/run.log exec >$LOGFILE 2>&1 # ****************************************** # 0. Process all samples independently # ****************************************** GVCF_INPUTS="" for SM in $SM_LIST; do RGID="rg_$SM" mkdir $WORKDIR/$SM cd $WORKDIR/$SM # ****************************************** # 1. Mapping reads with BWA-MEM, sorting # ****************************************** ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$RGID\tSM:$SM\tPL:$PL" \ -t $NT -K 10000000 $FASTA $FASTQ_FOLDER/${SM}_$FASTQ_1_SUFFIX \ $FASTQ_FOLDER/${SM}_$FASTQ_2_SUFFIX || { echo -n 'bwa error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -r $FASTA -o sorted.bam -t $NT --sam2bam -i - || \ { echo "Alignment failed"; exit 1; } # ****************************************** # 2. Metrics # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i sorted.bam \ --algo MeanQualityByCycle mq_metrics.txt --algo QualDistribution qd_metrics.txt \ --algo GCBias --summary gc_summary.txt gc_metrics.txt --algo AlignmentStat \ --adapter_seq '' aln_metrics.txt --algo InsertSizeMetricAlgo is_metrics.txt || \ { echo "Metrics failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o gc-report.pdf gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution -o qd-report.pdf qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle -o mq-report.pdf mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo -o is-report.pdf is_metrics.txt # ****************************************** # 3. Remove Duplicate Reads. It is possible # to remove instead of mark duplicates # by adding the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i sorted.bam --algo LocusCollector \ --fun score_info score.txt || { echo "LocusCollector failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i sorted.bam --algo Dedup \ --score_info score.txt --metrics dedup_metrics.txt deduped.bam || \ { echo "Dedup failed"; exit 1; } # ****************************************** # 2a. Coverage metrics # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i deduped.bam \ --algo CoverageMetrics coverage_metrics || { echo "CoverageMetrics failed"; exit 1; } # ****************************************** # 5. Base recalibration # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i deduped.bam --algo QualCal \ -k $KNOWN_DBSNP -k $KNOWN_MILLS -k $KNOWN_INDELS recal_data.table $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i deduped.bam -q recal_data.table \ --algo QualCal -k $KNOWN_DBSNP -k $KNOWN_MILLS -k $KNOWN_INDELS recal_data.table.post $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT --algo QualCal --plot \ --before recal_data.table --after recal_data.table.post recal.csv $SENTIEON_INSTALL_DIR/bin/sentieon plot QualCal -o recal_plots.pdf recal.csv # ****************************************** # 6b. HC Variant caller for GVCF # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i deduped.bam -q recal_data.table \ --algo Haplotyper -d $KNOWN_DBSNP --emit_conf=30 --call_conf=30 --emit_mode gvcf \ output-hc.g.vcf.gz || { echo "Haplotyper failed"; exit 1; } GVCF_INPUTS="$GVCF_INPUTS -v $WORKDIR/$SM/output-hc.g.vcf.gz" done # ****************************************** # Perform the joint calling # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA --algo GVCFtyper $GVCF_INPUTS \ -d $KNOWN_DBSNP $WORKDIR/output-jc.vcf.gz || { echo echo "GVCFtyper failed"; exit 1; } ``` <br> # 5 工具详细使用说明 ## 5.1 DRIVER二进制程序 DRIVER是用于执行生物信息学流程所有阶段的二进制程序。单次调用 driver 二进制文件可以运行多个算法;例如,指标统计阶段就是通过调用driver运行多个算法来实现的。 ### 5.1.1 DRIVER语法 DRIVER二进制文件的一般语法是: ``` sentieon driver OPTIONS--algo ALGORITHM ALGO_OPTION OUTPUT \ [--algo ALGORITHM2 ALGO_OPTION2 OUTPUT2] ``` 在同一次 driver 调用中运行多个算法的情况下,OPTIONS(选项)由所有算法共享。 该命令的参数包括: - `-t NUMBER_THREADS`:软件用于运行并行进程的计算线程数。此参数是可选的;如果省略,driver工具将使用服务器拥有的所有线程。 - `-r REFERENCE`:参考FASTA文件的位置。除了LocusCollector和不含共识序列的Dedup外,此参数在所有算法中都是必需的。 - `-i INPUT_FILE`:BAM 输入文件的位置。该参数可以使用多次,使软件使用多个输入文件,其结果与将输入 BAM 文件合并为一个 BAM 文件相同。Sentieon® 仅支持按坐标排序的 BAM 文件,且应包含带有 `SO:coordinate` 属性的 `@HD` 行。Sentieon® 还要求输入 BAM 文件的比对部分包含 `RG` 标签,且在文件头(header)中定义了具有匹配 ID 标签的对应 `@RG` 行。除 CollectVCMetrics、GVCFtyper、DNAModelApply、SVSolver、TNModelApply、VarCal、ApplyVarCal 和 TNfilter 外,所有算法都需要此参数。 - `-q QUALITY_RECALIBRATION_TABLE`:来自BQSR阶段的质量重校准表的位置,该表将在BQSR阶段和变异检测阶段作为输入使用。可以多次使用此参数使软件使用多个输入校准表;每个校准表将通过匹配读段的读组对BAM文件进行重校准。 - `--interval INTERVAL`:软件将使用的参考基因组中的区间。该参数可以使用多次,对所有区间的并集进行计算。INTERVAL可以指定为: - `CONTIG[:START-END]`:仅在对应的重叠群上进行计算。START和END是可选的数字,用于进一步缩小区间;二者均以 1 为起始基准,您可以输入以逗号分隔的多个重叠群列表。 - `BED_FILE`:包含区间的BED文件的路径。如果提供空文件,将不进行任何处理,等同于区间长度为0,同时也支持压缩的BGZF文件。 - `PICARD_INTERVAL_FILE`:包含区间的文件路径,遵循Picard区间标准。如果提供空文件,将不进行任何处理,等同于区间长度为0,同时也支持压缩的BGZF文件。 - `VCF_FILE`:包含变异记录的VCF文件路径,其基因组坐标将用作区间。同时也支持压缩的VCF.gz文件。 - `--interval_padding PADDING_SIZE`:为输入区间的边缘添加PADDING_SIZE大小的碱基填充。默认值为0。 - `--help`:显示帮助的选项。该选项可以与`--algo ALGORITHM`搭配使用,以显示特定算法的帮助。 - `--read_filter FILTER,OPTION=VALUE,OPTION=VALUE`:在应用算法之前执行读段的过滤或转换。请参考 *第5.1.3节* 获取有关可用过滤器及其功能的其他信息。 - `--temp_dir DIRECTORY`:确定临时文件的存储路径。默认值为运行命令的文件夹($PWD)。 - `--skip_no_coor`:确定是否跳过未映射的读段。 - `--cram_read_options decode_md=0`:CRAM输入选项,用于关闭输入CRAM中的NM/MD标签。 - `--replace_rg ORIG_RG="NEW_RG_STRING"`:修改下一个BAM输入文件的@RG读组标签,用新信息更新ORIG_RG;NEW_RG_STRING必须是有效且完整的字符串。此参数可以多次使用, 每次都会影响下一个 `-i` 指定的 BAM 输入。您可以查看 *第6.6节* 获取详细的使用示例。 该命令支持的算法包括: - LocusCollector、Dedup:用于去除重复序列阶段。 - Realigner:用于插入缺失重比对阶段。 - QualCal:用于碱基质量重校准阶段。 - MeanQualityByCycle、QualDistribution、GCBias、AlignmentStat、InsertSizeMetricAlgo、HsMetricAlgo、CoverageMetrics、BaseDistributionByCycle、QualityYield、WgsMetricsAlgo、SequenceArtifactMetricsAlgo:用于计算QC指标。 - Genotyper、Haplotyper:用于胚系变异检测分析。 - DNAscope、DNAModelApply:用于DNAscope胚系变异检测分析。 - DNAscope、SVsolver:用于DNAscope胚系结构变异分析。 - DNAscope、VariantPhaser:用于PacBio® HiFi®读段的DNAscope胚系变异检测分析。 - ReadWriter:用于输出碱基质量重校准后的BAM文件或合并多个BAM文件。 - VarCal、ApplyVarCal:用于VQSR阶段。 - GVCFtyper:用于胚系联合变异检测分析。 - TNsnv、TNhaplotyper、TNhaplotyper2、ContaminationModel、 OrientationBias、TNfilter:用于体细胞瘤瘤-正常配对或仅肿瘤样本分析。 - TNscope、TNModelApply:用于体细胞肿瘤-正常体细胞和结构变异分析。 - RNASplitReadsAtJunction:用于在接头(junctions)处分割RNA 读段的阶段。 - ContaminationAssessment:用于从BAM文件中识别样本间污染。 - CollectVCMetrics:用于计算变异检测后的指标。 ### 5.1.2 DRIVER算法 #### (1)LocusCollector LocusCollector算法收集将用于移除重复读段的读段信息。 LocusCollector算法的输入是BAM文件;其输出是指示哪些读段可能是重复序列。 LocusCollector算法需要以下ALGO_OPTION: - `--fun SCORE`:要使用的评分函数。SCORE的可能值为: - `score_info`:计算Dedup算法的读段对的评分。这是默认的评分函数。 - `--consensus`:设置此选项以开启共识去重。 - `--umi_tag TAG`:逻辑 UMI 标签,用于支持 UMI 条形码感知的去重。默认值为 None。 - `--umi_ecc_dist DISTANCE`:UMI条形码错误校正距离。将其设置为0以关闭条形码错误纠正。默认值为1。 - `--rna`:针对经STAR比对的RNA序列数据设置此选项。 #### (2)Dedup Dedup算法执行重复读段的标记/移除。 Dedup算法的输入是BAM文件;其输出是移除重复读段后的BAM文件。 Dedup算法需要以下ALGO_OPTION: - `--score_info LOCUS_COLLECTOR_OUTPUT`:LocusCollector命令检测的输出文件路径。 Dedup算法接受以下可选的ALGO_OPTION: - `--rmdup`:设置此选项以在输出BAM文件中移除重复的读段。如果未设置此选项,重复的读段将被标记,但不会从BAM文件中移除。 - `--cram_write_options compressor=[gzip|bzip2|lzma|rans]`:CRAM输出压缩选项。如果未定义,默认值为`compressor=gzip+rans`。 - `--cram_write_options version=[3.0|3.1]`:CRAM输出版本选项。如果未定义,默认为`version=3.0`。 - `--metrics METRICS_FILE`:包含去重阶段指标数据的输出文件的路径和文件名。 - `--optical_dup_pix_dist DISTANCE`:确定两个重复读段被视为光学重复的最大距离。默认值为100。 - `--bam_compression COMPRESSION_LEVEL[0-9]`:输出BAM文件的gzip压缩级别。默认值为6。 - `--output_dup_read_name`:使用此选项时,命令的输出将不是带有标记/移除重复的BAM文件,而是被标记为重复的读段名称列表。 - `--dup_read_name DUPLICATE_READ_NAME_FILE`:使用此输入时,DUPLICATE_READ_NAME_FILE中包含的所有读段将被标记为重复,无论它们是主要还是非主要读段。 #### (3)Realigner Realigner算法执行插入缺失(indel)重新比对。 Realigner算法的输入是BAM文件;其输出是重新比对后的BAM文件。 Realigner算法接受以下可选的ALGO_OPTION: - `-k KNOWN_SITES`:用作已知位点集的VCF文件路径。已知位点将用于帮助识别可能需要重新比对的位点;仅使用文件中的indel变异。您可以通过指定多个文件并重复 `-k KNOWN_SITES` 选项来包含多组已知位点。 - `--interval_list INTERVAL`:参考基因组中的区间,将用于计算重比对目标。仅考虑单个输入的 INTERVAL:如果您重复使用 `--interval_list` 选项,则仅考虑最后一个 INTERVAL。INTERVAL可以指定为: - `BED_FILE`: 包含区间的BED文件的路径。 - `PICARD_INTERVAL_FILE`: 包含区间的文件的路径,遵循Picard区间标准。 - `--bam_compression COMPRESSION_LEVEL[0-9]`:输出BAM文件的gzip压缩级别。默认值为6。 - `--cram_write_options compressor=[gzip|bzip2|lzma|rans]`:CRAM输出压缩选项。如果未定义,默认为`compressor=gzip+rans`。 - `--cram_write_options version=[3.0|3.1]`:CRAM输出版本选项。如果未定义,默认为version=3.0。 #### (4)QualCal QualCal算法用于计算执行碱基质量分数重校准(BQSR)所需的重校准表。QualCal算法还应用重校准来计算创建报告所需的数据,并创建生成报告所需的数据。 重校准的计算依赖于读组的平台(PL)标签;QualCal算法支持以下平台:ILLUMINA、ION_TORRENT、LS454、PACBIO、COMPLETE_GENOMICS、DNBSEQ。目前尚未实现对SOLID平台测序数据的支持。 QualCal算法的输入是BAM文件;其输出是重校准表或包含创建报告所需数据的csv文件。 QualCal算法接受以下可选的ALGO_OPTION: - `-k KNOWN_SITES`:用作已知位点集的 VCF 文件路径。已知位点将用于确保已知位点不会因将真实变异错误识别为测序过程中的错误,而人为地获得较低的质量分数。您可以通过指定多个文件并重复使用 `-k KNOWN_SITES` 选项来包含多组已知位点。我们强烈建议使用尽可能多的已知位点,否则重校准过程会将变异位点视为测序错误。 - `--plot`:指示该命令是否用于生成创建报告所需的数据。 - `--cycle_val_max`:周期协变量允许的最大周期值。 - `--before RECAL_TABLE`:先前计算的重校准表的路径;它将用于应用重校准。 - `--after RECAL_TABLE.POST`:先前计算的应用重校准表结果的路径;它将用于计算创建报告所需的数据。 #### (5)MeanQualityByCycle MeanQualityByCycle 算法用于计算每个测序周期的平均碱基质量分数。 MeanQualityByCycle 算法的输入是一个 BAM 文件;其输出的是指标数据。 MeanQualityByCycle 算法不接受任何 ALGO_OPTION。 #### (6)QualDistribution QualDistribution 算法用于计算具有特定碱基质量分数的碱基数量。 QualDistribution 算法的输入是一个 BAM 文件;其输出的是指标数据。 QualDistribution 算法不接受任何 ALGO_OPTION。 #### (7)GCBias GCBias 算法用于计算参考基因组和样本中的 GC 偏好性。 GCBias 算法的输入是一个 BAM 文件;其输出的是指标数据。 GCBias 算法接受以下可选的 ALGO_OPTION: - `--summary SUMMARY_FILE`:汇总 GC 偏好性指标的输出文件的位置和文件名。 - `--accum_level LEVEL`:确定累积级别。LEVEL 的可能值包括 ALL_READS、SAMPLE、LIBRARY、READ_GROUP。默认值是 ALL_READS。 - `--also_ignore_duplicates`:确定输出指标是否使用唯一的非重复读段进行计算。 - `--is_bisulfite_seq`:设置此选项表明输入的BAM文件有亚硫酸氢盐测序的读段组成。 #### (8)HsMetricAlgo HsMetricAlgo 算法用于计算样本的杂交选择特定指标以及参考基因组的 AT/GC 含量脱落指标。 HsMetricAlgo 算法的输入是一个 BAM 文件;其输出的是指标数据。 HsMetricAlgo 算法需要以下 ALGO_OPTION: - `--targets_list TARGETS_FILE`:包含目标位置的间隔列表输入文件的路径和文件名。 - `--baits_list TARGETS_FILE`:包含所使用诱饵位置的间隔列表输入文件的路径和文件名。 - `--clip_overlapping_reads`:确定在计算过程中是否切除重叠的读段。 - `--min_map_qual QUALITY`:确定所使用读段的过滤质量。任何映射质量低于 QUALITY 的读段都将被过滤掉。 - `--min_base_qual QUALITY`:确定变异检测中使用的碱基的过滤质量。任何质量低于 QUALITY 的碱基都将被忽略。 - `--coverage_cap COVERAGE`:确定直方图中使用的最大覆盖度限制。 #### (9)AlignmentStat AlignmentStat 算法用于计算读段比对的统计数据。 AlignmentStat 算法的输入是一个 BAM 文件;其输出的是指标数据。 AlignmentStat 算法接受以下可选的 ALGO_OPTION: - `--adapter_seq SEQUENCE_LIST`:测序中使用的接头序列,以逗号分隔的列表形式提供。默认值为默认的 Illumina 接头列表。 - `--is_bisulfite_seq`:设置此选项表明输入的BAM文件由亚硫酸氢盐测序的读段组成。 #### (10)InsertSizeMetricAlgo InsertSizeMetricAlgo 算法用于计算插入片段大小的统计分布。 InsertSizeMetricAlgo 算法的输入是一个 BAM 文件;其输出的是指标数据。 InsertSizeMetricAlgo 算法接受以下可选的 ALGO_OPTION: - `--deviation DEVIATION`:在直方图被裁剪之前的最大标准偏差倍数。默认值为 10.0。 - `--hist_width HISTOGRAM_WIDTH`:设置 HISTOGRAM_WIDTH 以覆盖直方图尾部的自动截断。默认值为 0。 - `--min_read_ratio RATIO`:要包含在直方图中的读取类别(read category)的最小读取比例。默认值为 0.05。 - `--se_tag TAG`:在直方图中包含带有该 TAG的单端读段。默认值为 MI。 #### (11)CoverageMetrics CoverageMetrics 算法计算 BAM 文件的深度覆盖度。如果在 driver 级别包含 `--interval` 选项,则覆盖度将按区间聚合;如果不包含 `--interval` 选项,则参考基因组中所有碱基的每个区间聚合将按重叠群进行,且每个位点的覆盖度输出文件大小约为 60GB。如果包含 RefSeq 文件,则覆盖度将按基因聚合。 CoverageMetrics 算法的输入是 BAM 文件,建议使用去重复后的 BAM 文件进行此分析;其输出是按分区、聚合和输出组织的指标数据的文件。可能的输出包括: - summary:包含深度数据。 - statistics:包含具有特定深度的位点直方图。 - cumulative_coverage_counts:包含深度大于 x 的位点直方图。 - cumulative_coverage_proportions:包含深度大于 x 的位点的归一化直方图。 当输出名称为 OUTPUT时的输出文件示例: - OUTPUT:没有分区的每个位点覆盖度。 - OUTPUT.sample_summary:针对分区组样本的汇总,对所有碱基进行聚合。 - OUTPUT.library_interval_statistics:针对分区组文库的统计数据,按区间聚合。 CoverageMetrics 算法接受以下可选的 ALGO_OPTION: - `--partition PARTITION_GROUP`:确定如何分区数据。可能的值是 readgroup 或 RG 属性的逗号分隔组合,即 sample、platform、library、center。默认值是 "sample"。可以通过重复该选项包含多个分区组,每个输出文件将为每个选项创建一次。 - `--partition PARTITION\_GROUP`:确定如何对数据进行分区。可能的值为 readgroup,或者由 RG 属性(即 sample、platform、library、center)组成的以逗号分隔的组合。默认值为 "sample"。您可以通过重复使用 `--partition` 选项来包含多个分区组,每个输出文件将针对每个 `--partition` 选项创建一次。 - `--gene_list REFSEQ_FILE`:用于将 CoverageMetrics 算法的结果聚合到基因级别的 RefSeq 文件的路径。 - 过滤选项: - `--min_map_qual MAP_QUALITY`:确定使用读段的过滤质量。任何比对质量低于 QUALITY 的读段将被过滤掉。 - `--max_map_qual MAP_QUALITY`:确定使用读段的过滤质量。任何比对质量高于 QUALITY 的读段将被过滤掉。 - `--min_base_qual QUALITY`:确定使用碱基的过滤质量。任何质量低于 QUALITY 的碱基将被忽略。 - `--max_base_qual QUALITY`:确定使用碱基的过滤质量。任何质量高于 QUALITY 的碱基将被忽略。 - `--cov_thresh THRESHOLD`:在聚合中添加覆盖度大于阈值的碱基百分比。可以通过重复使用 `--cov_thresh`参数包含多个阈值。 - 省略输出选项: - `--omit_base_output`:跳过没有分区的每个位点覆盖度的输出。当您不使用区间时,此选项可用于节省空间。 - `--omit_sample_stat`:跳过对所有碱基聚合的汇总结果(`_summary`)的输出。 - `--omit_locus_stat`:跳过所有直方图文件(包括 `_cumulative_coverage_counts` 和 `_cumulative_coverage_proportions`)的输出。 - `--omit_interval_stat`:跳过所有区间统计文件(`_interval_statistics`)的输出。 - `--count_type TYPE`:确定如何处理来自同一片段的重叠读段。可能的选项有: - `0`:即使重叠读段来自同一片段,也对其进行计数。这是默认值。 - `1`:对重叠读段进行计算。 - `2`:仅当片段中的读段具有一致的碱基时,才对重叠读段进行计数。 - `--print_base_counts`:在没有分区的每个位点覆盖度输出中包含 "AGCTND" 的数量。 - `--include_ref_N`:在参考基因组设置为 N的位点包含覆盖度数据。 - `--ignore_del_sites`:忽略存在缺失的位点的覆盖率数据。 - `--include_del`:此参数将与其他参数交互如下: - 如果 `ignore_del_sites` 关闭,则将删除计入深度 - 如果 `print_base_counts` 开启,则包括 'D' 的数量 - `--histogram_scale [log/linear] --histogram_low MIN_DEPTH --histogram_high MAX_DEPTH --histogram_bin_count NUM_BINS`:确定直方图的比例类型、bin 和大小。默认值为 log、1、500、499。 #### (12)CollectVCMetrics CollectVCMetrics 算法收集与输入 VCF 文件中存在的变异相关的指标。 CollectVCMetrics 算法的输入是一个 VCF 文件和一个 DBSNP 文件;其输出的是一对包含 VCF 文件中变异信息的文件。 CollectVCMetrics 算法需要以下 ALGO_OPTION: - `-d dbSNP_FILE`:单核苷酸多态性数据库(dbSNP)的位置。仅支持单个文件。 - `-v INPUT`:将用于计算度量指标的 VCF 文件的位置。仅支持单个文件。 #### (13)BaseDistributionByCycle BaseDistributionByCycle 算法计算每个测序周期的核苷酸分布。 BaseDistributionByCycle 算法的输入是一个 BAM 文件;其输出的是指标数据。 BaseDistributionByCycle 算法接受以下可选的 ALGO_OPTION: - `--aligned_reads_only`:确定是否仅基于已比对的读段计算碱基分布。 - `--pf_reads_only`:确定是否仅基于通过过滤 (PF) 的读段计算碱基分布。 #### (14)QualityYield QualityYield 算法收集与通过质量阈值和 Illumina 特定过滤器的读段相关的指标。 QualityYield 算法的输入是一个 BAM 文件;其输出的是指标数据。 QualityYield 算法接受以下可选的 ALGO_OPTION: - `--include_secondary`:确定计算中是否包含次级比对的碱基。 - `--include_supplementary`:确定计算中是否包含补充比对的碱基。 #### (15)WgsMetricsAlgo WgsMetricsAlgo 算法收集与全基因组测序(WGS)实验的覆盖度和性能相关的指标。 WgsMetricsAlgo 算法的输入是一个 BAM 文件;其输出的是指标数据。 WgsMetricsAlgo 算法接受以下可选的 ALGO_OPTION: - `--min_map_qual QUALITY`:确定计算中所用读段的过滤质量。任何质量低于 QUALITY的读段将被忽略。默认值为 20。 - `--min_base_qual QUALITY`:确定计算中所用碱基的过滤质量。任何质量低于 QUALITY的碱基将被忽略。默认值为 20。 - `--coverage_cap COVERAGE`:确定直方图的最大覆盖度限制。任何覆盖度高于 COVERAGE的位置其覆盖度都将被设为COVERAGE。 - `--sample_size SIZE`:确定用于理论杂合敏感性采样的样本大小。默认值为 10000。 - `--include_unpaired`:确定是否计算未配对的读段和一端未比对的配对读段。 - `--base_qual_histogram`:确定是否报告碱基质量直方图。 #### (16)SequenceArtifactMetricsAlgo SequenceArtifactMetricsAlgo 算法收集用于量化单碱基测序伪影和 OxoG 伪影的指标。 SequenceArtifactMetricsAlgo 算法的输入是一个 BAM 文件;其输出是指标数据。 SequenceArtifactMetricsAlgo 算法接受以下可选的 ALGO_OPTION: - `--dbsnp FILE`:单核苷酸多态性数据库 (dbSNP) 的位置,用于排除已知多态性位点周围的区域。仅支持单个文件。 - `--min_map_qual QUALITY`:确定用于计算的读段的过滤质量。任何质量低于 QUALITY 的读段将被忽略。默认值为 30。 - `--min_base_qual QUALITY`:确定用于计算的碱基的过滤质量。任何质量低于 QUALITY 的碱基将被忽略。默认值为 20。 - `--include_unpaired`:确定是否计算未配对的读段和一端未比对的配对读段。 - `--include_duplicates`:确定是否计算重复的读段。 - `--include_non_pf_reads`:确定是否计算非 PF 的读段。 - `--min_insert_size ISIZE`:确定用于计算的读段的过滤插入片段大小。任何插入片段大小小于 ISIZE 的读段将被忽略。默认值为 60。 - `--max_insert_size ISIZE`:确定用于计算的读段的过滤插入片段大小。任何插入片段大小大于 ISIZE 的读段将被忽略。默认值为 600。 - `--tandem_reads`:确定配对读段是否在同一条链上测序。 - `--context_size SIZE`:确定在每侧包含的上下文碱基数量。默认值为 1。 #### (17)Genotyper Genotyper算法用于执行统一基因型变异检测。 Genotyper算法的输入是BAM文件;输出的是一个VCF文件。 Genotyper算法接受以下可选的ALGO_OPTION: - `--annotation 'ANNOTATION_LIST'`:确定将添加到输出VCF的其他注释。使用逗号分隔的列表来启用或禁用注释。包括'none'以删除默认注释;在注释前加感叹号(!)以禁用特定注释。有关支持的注释,请参见 *第6.2节*。 - `-d dbSNP_FILE`:用于标记已知变异的单核苷酸多态性数据库(dbSNP)的路径。仅支持单个文件。 - `--var_type VARIANT_TYPE`:确定将检测哪些变异类型;VARIANT_TYPE的可能值为: - SNP :仅检测单核苷酸多态性。这是默认行为。 - INDEL:仅检测插入/缺失。 - both :同时检测SNP和INDEL。 - `--call_conf CONFIDENCE`:确定检测变异的变异质量阈值。质量低于CONFIDENCE的变异将被移除。 - `--emit_conf CONFIDENCE`:确定输出变异的变异质量阈值。质量低于CONFIDENCE的变异将不会被添加到输出的VCF文件中。 - `--emit_mode MODE`:确定将输出哪些检测结果。MODE的可能值为: - variant:仅在置信的变异位点输出检测结果。这是默认行为。 - confident:在置信的变异位点或置信的参考位点输出检测结果。 - all:输出所有检测结果,无论其置信度如何。 - `--min_base_qual QUALITY`:确定变异检测中使用的碱基过滤质量。任何质量低于QUALITY的碱基将被忽略。默认值为17。 - `--ploidy PLOIDY`:确定正在处理的样本的倍性数。默认值为2。 - `--given GIVEN_VCF`:仅使用GIVEN_VCF中提供的变异执行变异检测。检测将仅评估文件中提供的位点和等位基因,并且仅当变异的FILTER列为PASS或.时才进行。 - `--genotype_model MODEL`:确定用于基因型分型和QUAL计算的模型。MODEL可以是coalescent使用基于群体遗传学中的合并理论的"所谓精确模型",或multinomial使用假设变异是独立的简化模型;默认值是coalescent。 #### (18)Haplotyper Haplotyper算法用于执行基于单倍型的变异检测。 Haplotyper算法的输入是BAM文件;输出的是一个VCF文件。 Haplotyper算法接受以下可选的ALGO_OPTION: - `--annotation 'ANNOTATION_LIST'`:确定将添加到输出VCF的其他注释。使用逗号分隔的列表来启用或禁用注释。包括'none'以删除默认注释;在注释前加感叹号(!)以禁用特定注释。有关支持的注释,请参见 *第6.2节*。 - `-d dbSNP_FILE`:用于标记已知变异的单核苷酸多态性数据库(dbSNP)的位置。仅支持单个文件。 - `--call_conf CONFIDENCE`:确定检测变异的变异质量阈值。质量低于CONFIDENCE的变异将被移除。当--emit_mode为gvcf时,此选项将被忽略。 - `--emit_conf CONFIDENCE`:确定输出变异的变异质量阈值。质量低于CONFIDENCE的变异将不会添加到输出VCF文件中。当--emit_mode为gvcf时,此选项将被忽略。 - `--emit_mode MODE`:确定将输出哪些检测结果。MODE的可能值为: - variant:仅在置信的变异位点输出检测结果。这是默认行为。 - confident:在置信的变异位点或置信的参考位点输出检测结果。 - all:输出所有检测结果,无论其置信度如何。 - gvcf:输出联合检测所需的附加信息。如果要使用GVCFtyper算法执行联合检测,则需要此选项。 - `--gq_bands LIST_OF_BANDS`:确定用于压缩具有相似基因型质量(GQ)的变异的波段,这些变异将在GVCF输出文件中作为单个VCF记录输出。LIST_OF_BANDS是一个逗号分隔的波段列表,其中每个波段由START-END/STEP定义。默认值为1-60,60-99/10,99。 - `--min_base_qual QUALITY`:确定用于变异检测的碱基的过滤质量。任何质量低于QUALITY的碱基将被忽略。默认值为10。 - `--pcr_indel_model MODEL`:PCR indel 模型,用于或多或少激进地剔除假阳性 indel。可能的 MODEL包括:NONE(用于无 PCR 样本),以及 HOSTILE、AGGRESSIVE 和CONSERVATIVE (按激进程度递减排序)。默认值为 CONSERVATIVE。 - `--phasing [1/0]`:在使用emit_mode GVCF时启用或禁用输出中的相位的标记。默认值为1(开启),此标志在使用GVCF以外的emit_mode时无影响。相位仅针对二倍体样本进行计算。 - `--ploidy PLOIDY`:确定正在处理的样本的倍性数。默认值为2。 - `--prune_factor FACTOR`:局部组装中的最小剪枝因子;支持k-mer数量少于FACTOR的路径将从图中剪除。默认值为2。 - `--trim_soft_clip`:确定是否应从变异检测中排除读段中的软剪切碱基。仅建议在处理RNA读段时使用此参数。 - `--given GIVEN_VCF`:仅使用 GIVEN_VCF中提供的变异执行变异检测。检测将仅评估文件中提供的位点和等位基因,且仅当变异的 FILTER 列为 PASS 或 . 时才进行。此选项不能与 --emit_mode gvcf连用。 - `--bam_output OUTPUT_BAM`:输出一个 BAM 文件,其中包含变异检测执行局部重组后修改过的读段。此选项应仅与较小的 BED 文件连用,用于故障排查目的。 - `--genotype_model MODEL`:确定用于基因型分型和QUAL计算的模型。MODEL可以是coalescent使用基于群体遗传学中的合并理论的"所谓精确模型",或multinomial使用假设变异是独立的简化模型;默认值是coalescent。 #### (19)ReadWrite ReadWriter算法将应用碱基质量分数重校准(BQSR)后的结果输出到文件中。 ReadWriter算法还可以合并BAM文件,或将它们转换为CRAM文件。 ReadWriter算法的输入是一个或多个BAM文件和一个或多个重校准表;其输出是重校准后的BAM文件。如果输出文件扩展名为CRAM,将生成CRAM文件。如果使用了多个输入文件,输出文件将是合并所有文件的结果。 ReadWriter算法接受以下可选的ALGO_OPTION: - `--bam_compression COMPRESSION_LEVEL[0-9]`:输出BAM文件的gzip压缩级别。默认值为6。 - `--cram_write_options compressor=[gzip|bzip2|lzma|rans]`:CRAM输出压缩选项。如果未定义,默认为`compressor=gzip+rans`。 - `--cram_write_options version=[3.0|3.1]`:CRAM输出版本选项。如果未定义,默认为`version=3.0`。 - `--output_mapq_filter min_map_qual=MIN_QUAL,max_map_qual=MAX_QUAL`:基于比对质量过滤读段;ReadWriter 仅输出比对质量大于或等于MIN_QUAL且小于或等于 MAX_QUAL的读段。 - `--output_flag_filter MASK`:VALUE[,VALUE,VALUE...]:基于标志过滤读段。MASK是应考虑的位集,VALUE是以逗号分隔的FLAG列表,表示保留读段所需设置的标志,如果MASK中的所有位都需要未设置,则为0。您可以包含此选项的多个实例,它们将按命令行中的顺序依次应用。一些示例: - 仅输出`PROPER_PAIR`的读段,您可以使用` --output_flag_filter PROPER_PAIR:PROPER_PAIR `或 `--output_flag_filter 0x2:0x2` - 要输出除带有UNMAP标志的未比对读段之外的所有读段,您可以使用 `--output_flag_filter UNMAP:0 `或 `--output_flag_filter 0x4:0` - 写入一个不包含重复读段或未比对读段 的 BAM 文件,您可以使用两个顺序应用的选项:`--output_flag_filter UNMAP:0 --output_flag_filter DUP:0`。这将首先仅允许取消设置UNMAP标志的读段,然后仅允许取消设置DUP标志的读段。 - 要写入一个不包含两个配对读段都未比对的BAM文件,您可以使用 `--output_flag_filter UNMAP+MUNMAP:0,UNMAP,MUNMAP`,只要不同时设置UNMAP 和MUNMAP,就允许该读段。 - 更复杂的情况可以借助真值表构建,其中MASK是您想考虑的所有位的总和,而VALUE是每种不同条件下设置的那些位的总和。例如,要查找两个非二次和非补充读段具有相同方向(F1F2或R1R2)的读段对(暗示结构变异),您可以创建如下表格: 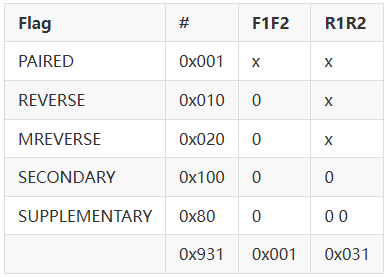 对应的选项将是 `--output_flag_filter 0x931:0x001,0x031 ` 或更复杂的形式 `--output_flag_filter PAIRED+SECONDARY+SUPPLEMENTARY+DUP+REVERSE+MREVERSE:PAIRED+REVERSE+MREVERSE,PAIRED+REVERSE+MREVERSE` 下表是标志定义及其命名规定的提示: 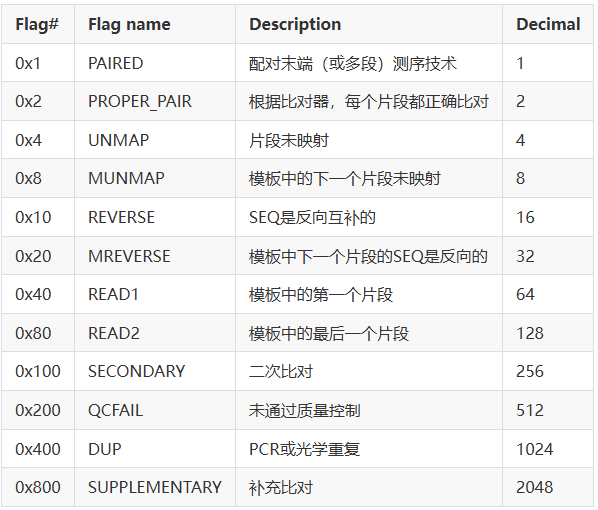 我们建议在执行变异检测命令的同时运行ReadWriter算法,以减少系统开销。 #### (20)GVCFtyper GVCFtyper算法执行多个样本的联合变异检测,前提是每个单个样本之前已使用Haplotyper算法和--emit_mode gvcf选项进行处理。 GVCFtyper算法在driver 级别除参考文件外没有输入,其输出的是一个包含所有样本联合检测变异的VCF文件。 GVCFtyper算法需要以下ALGO_OPTION: - `-v INPUT`:使用额外选项`--emit_mode gvcf`对单个样本执行变异检测算法所得的GVCF文件的路径。您可以通过指定多个文件并重复`-v INPUT`选项来包含来自多个样本的GVCF文件。您可以使用经bgzip压缩并建立索引的VCF文件。 - 或者,您可以在命令末尾的输出文件之后输入GVCF文件列表。因此,以下两条命令是等效的: ``` sentieon driver -r REFERENCE --algo GVCFtyper \ -v s1_VARIANT_GVCF -v s2_VARIANT_GVCF -v s3_VARIANT_GVCF VARIANT_VCF sentieon driver -r REFERENCE --algo GVCFtyper \ VARIANT_VCF s1_VARIANT_GVCF s2_VARIANT_GVCF s3_VARIANT_GVCF ``` - 您可以使用以下命令从文本文件读取GVCF文件列表: ``` gvcf_argument="" while read -r line; do gvcf_argument=$gvcf_argument" -v $line" done < "list_of_gvcfs" sentieon driver -r REFERENCE --algo GVCFtyper $gvcf_argument output-joint.vcf ``` - 您还可以使用以下利用标准输入管道(-)的命令从文本文件读取GVCF文件列表: ``` cat list_of_gvcfs | sentieon driver -r REFERENCE --algo GVCFtyper output-joint.vcf - ``` - 您可以使用以下命令输入特定文件夹中的所有文件: ``` sentieon driver -r REFERENCE --algo GVCFtyper output-joint.vcf sample*.g.vcf ``` GVCFtyper算法接受以下可选的ALGO_OPTION: - `--annotation 'ANNOTATION_LIST'`:确定将添加到输出VCF的其他注释。使用逗号分隔的列表来启用或禁用注释。包括'none'以删除默认注释;在注释前加感叹号(!)以禁用特定注释。有关支持的注释,请参见 *第6.2节*。 - `-d dbSNP_FILE`:用于标记已知变异的单核苷酸多态性数据库(dbSNP)的路径。仅支持单个文件。 - `--call_conf CONFIDENCE`:确定检测变异的变异质量阈值。质量低于CONFIDENCE的变异将被移除。默认值为30。 - `--emit_conf CONFIDENCE`:确定输出变异的变异质量阈值。质量低于CONFIDENCE的变异将不会添加到输出VCF文件中。默认值为30。 - `--emit_mode MODE`:确定将输出哪些检测结果。MODE的可能值为: - variant:仅在置信的变异位点输出检测解惑。这是默认行为。 - confident:在置信的变异位点或置信的参考位点输出检测结果。 - all:输出所有检测结果,无论其置信度如何。 - `--max_alt_alleles NUMBER` :最大变异等位基因数量。默认值为100。 - `--genotype_model MODEL`:确定用于基因型分型和QUAL计算的模型。MODEL可以是coalescent使用基于群体遗传学中的合并理论的"所谓精确模型",或multinomial使用假设变异是独立的简化模型;默认值是coalescent。 #### (21)VarCal VarCal算法用于计算变异质量分数重校准(VQSR)。VQSR为单个变异检测分配一个经过良好校准的概率分数,以便在确定最可能的变异时能够更准确地控制。为此,VQSR使用高度可信的已知位点来构建重校准模型,并确定检测位点为真的概率。有关该算法的更多信息,您可以查看http://gatkforums.broadinstitute.org/discussion/39/variant-quality-score-recalibration-vqsr 。有关VQSR中推荐使用的资源信息,您可以查看 https://www.broadinstitute.org/gatk/guide/article?id=1259 。 VarCal算法在driver级别除参考文件外没有输入,其输出的是一个包含与VQSR相关的额外注释的重校准文件。 VarCal算法需要以下ALGO_OPTION: - `-v INPUT`:变异检测算法生成的VCF文件的路径;您可以使用经bgzip压缩并建立索引的VCF文件。 - `--tranches_file TRANCHES_FILE`:包含检测集划分为质量等级的文件的位路径和文件名。 - `--annotation ANNOTATION`:确定在重校准期间将使用的注释。您可以通过重复使用该选项在优化中包含多个注释。您可以使用原始变异检测文件中存在的所有注释。 - `--resource RESOURCE_FILE --resource_param PARAM`:用作VQSR中训练/真实资源的VCF文件的位置,后跟确定该文件如何使用的参数。您可以通过指定多个文件并重复使用该选项来包含多个资源集。PARAM参数遵循以下语法: ``` LABEL,known=IS_KNOWN,training=IS_TRAIN,truth=IS_TRUTH,prior=PRIOR ``` - LABEL是资源的描述性名称。 - IS_KNOWN可以是true或false,确定资源中包含的位点是否将用于分层输出指标。 - IS_TRAIN可以是true或false,确定资源中包含的位点是否将用于训练重校准模型。 - IS_TRUTH可以是true或false,确定资源中包含的位点是否将被视为真实位点。 - PRIOR是一个值,反映了您对该资源作为真实集的可靠性的置信度。 VarCal算法接受以下可选的ALGO_OPTION: - `--srand RANDOM_SEED`:确定用于随机数生成的种子。您可以将`RANDOM_SEED`设置为0,软件将使用您计算机的随机种子。为了生成确定性结果,您应使用非零`RANDOM_SEED`并将`NUMBER_THREADS`设置为1。 - `--var_type VARIANT_TYPE`:确定将对哪些变异类型进行重校准;`VARIANT_TYPE`的可能值为: - SNP:仅对单核苷酸多态性进重校准。这是默认行为。 - INDEL:仅对插入-缺失进行重校准。 - BOTH:同时对SNP和INDEL进行重校准。不应使用此设置,因为应该对SNP和INDEL独立执行VQSR。 - `--tranche TRANCH_THRESHOLD`:每个等级的归一化质量阈值;`TRANCH_THRESHOLD`数字是0到100之间的数字。允许多个选项实例,这将创建与阈值一样多的等级。默认值为90、99、99.9和100。 - `--max_gaussians MAX_GAUSS`:确定将用于正重校准模型的最大的高斯分布数量。SNP的默认值为8,INDEL的默认值为4。 - `--max_neg_gaussians MAX_GAUSS`:确定将用于负重校准模型的最大的高斯分布数量。默认值为2。 - `--max_iter MAX_ITERATIONS`:确定期望最大化(EM)优化的最大迭代次数。默认值为150。 - `--max_mq MAPQ`: 指示数据中的最大MQ,将用于执行MQ的logit jitter转换,使其分布更接近高斯分布。 - `--aggregate_data AGREGATE_VCF`:包含从其他类似样本检测的变异的额外VCF文件的路径;这些额外数据将增加统计模型校准的有效样本量。允许包含该选项的多个实例。 - `--plot_file PLOT_FILE`:包含从VarCal算法生成报告所需数据的临时文件的位置。 #### (22)ApplyVarCal ApplyVarCal算法将来自VQSR的输出信息与原始变异信息相结合。 ApplyVarCal算法在driver级别除参考文件外没有输入;其输出的是一个包含 VQSR 额外注释的原始 VCF 文件的副本。 ApplyVarCal算法需要以下ALGO_OPTION: - `-v INPUT`:变异检测算法生成的VCF文件的位置。它应该与VarCal算法中使用的文件相同;您可以使用经bgzip压缩并建立索引的VCF文件。 - `--recal VARIANT_RECAL_DATA`:VarCal算法输出的重校准文件的位置。 - `--tranches_file TRANCHES_FILE`:VarCal算法输出的分级文件的位置。 - `--var_type VARIANT_TYPE`:确定重校准的变异类型。此选项应与VarCal算法中使用的选项保持一致。 或者,您可以使用以下选项输入多个VQSR模型所需信息的逗号分隔列表;此选项允许您在单个命令行中应用SNP和INDEL模式。该选项的语法是: ``` --vqsr_model var_type=VARIANT_TYPE,\ recal=VARIANT_RECAL_DATA,\ tranches_file=TRANCHES_FILE,\ sensitivity=SENSITIVITY ``` ApplyVarCal算法接受以下可选的ALGO_OPTION: - `--sensitivity SENSITIVITY`:确定对可用真实位点的敏感性;只有阈值大于敏感性的等级才会包含在重校准中。我们建议您使用包含在VarCal算法的等级阈值列表中的敏感性数字;这将减少舍入问题。默认值为NULL,即不应用等级过滤,仅应用LOW_VQSLOD过滤器。 #### (23)TNsnv TNsnv算法使用Genotyper算法对肿瘤-正常配对样本或肿瘤及正常样本面板数据执行体细胞变异检测。 TNsnv算法的输入是BAM文件;其输出的是一个VCF文件。 TNsnv算法需要以下ALGO_OPTION: - `--tumor_sample SAMPLE_NAME`:BAM文件中肿瘤样本的SM标签名称。 根据运行模式的不同,TNsnv算法可能需要以下ALGO_OPTION: - `--normal_sample SAMPLE_NAME`:BAM文件中正常样本的SM标签名称。 - `--detect_pon`:表示您正在使用TNsnv算法创建将成为正常样本面板一部分的VCF文件。 - `--cosmic COSMIC_VCF`:用于创建正常样本面板文件的体细胞突变目录(COSMIC)VCF文件的位置。仅支持单个文件。 - `--pon PANEL_OF_NORMAL_VCF`:包含在正常样本面板分析中检测到的变异的文件位置,用于去除假阳性。仅支持单个文件。 TNsnv算法接受以下可选的ALGO_OPTION: - `--dbsnp dbSNP_FILE`:单核苷酸多态性数据库(dbSNP)的位置。dbSNP中的变异更有可能被标记为胚系变异,因为它们需要更多证据证明在正常样本中不存在。仅支持单个文件。 - `--call_stats_out CALL_STATS_FILE`:包含体细胞变异检测检测统计信息的文件的输出位置和文件名。 - `--stdcov_out COVERAGE_FILE`:包含标准覆盖度的wiggle文件的路径和文件名。 - `--tumor_depth_out TUMOR_DEPTH_FILE`:包含肿瘤样本读段深度的wiggle文件的路径和文件名。 - `--normal_depth_out NORMAL_DEPTH_FILE`:包含正常样本读段深度的wiggle文件的路径和文件名。 - `--power_out POWER_FILE`:效检力(power)文件的路径和文件名。 - `--min_base_qual QUALITY`:确定变异检测中使用的碱基质量过滤。任何质量低于QUALITY的碱基将被忽略。默认值为5。 - `--min_init_tumor_lod NUMBER`:初始通过检测变异时的最小肿瘤对数比值。默认值为4。 - `--min_tumor_lod NUMBER`:最终变异检测时的最小肿瘤对数比值。默认值为6.3。 - `--min_normal_lod NUMBER`:用于检查肿瘤变异是否非正常样本变异的最小正常对数比值。默认值为2.2。 - `--contamination_frac NUMBER`:来自其他样本的污染比例估计值。默认值为0.02。 - `--min_cell_mutation_frac NUMBER`:携带突变的细胞的最小比例。默认值为0。 - `--min_strand_bias_power NUMBER`:检测链偏好性的最小功效。默认值为0.9。 - `--min_dbsnp_normal_lod NUMBER`:在dbSNP位点检测正常非变异的最小对数比值。默认值为5.5。 - `--min_normal_allele_frac NUMBER`:在正常样本中考虑的最小等位基因比例;当正常样本被肿瘤样本污染时,此参数非常有用。默认值为0。 - `--min_tumor_allele_frac NUMBER`:肿瘤样本中的最小等位基因频率。默认值为0.005。 - `--max_indel NUMBER`:允许的附近最大插入/缺失事件数量。默认值为3。 - `--max_read_clip_frac NUMBER`:读段中软/硬裁剪碱基的最大比例。默认值为0.3。 - `--max_mapq0_frac NUMBER`:比对质量为 0 的读段最大占比,用于确定差比对区域。默认值为0.5。 - `--min_pir_median NUMBER`:最小读段位置中位数。默认值为10。 - `--min_pir_mad NUMBER`:最小读段位置中位数绝对偏差。默认值为3。 - `--max_alt_mapq NUMBER`:变异等位基因比对质量得分的最大值。默认值为20。 - `--max_normal_alt_cnt NUMBER`:正常堆积中变异等位基因计数的最大值。默认值为2。 - `--max_normal_alt_qsum NUMBER`:正常堆积中变异等位基因质量得分总和的最大值。默认值为20。 - `--max_normal_alt_frac NUMBER`:正常堆积中变异等位基因的最大比例。默认值为0.03。 - `--power_allele_frac NUMBER`:用于效检力计算的等位基因频率。默认值为0.3。 #### (24)TNhaplotyper TNhaplotyper算法使用Haplotyper算法对肿瘤-正常配对样本或肿瘤和正常样本面板数据执行体细胞变异检测。 TNhaplotyper算法的输入是BAM文件;其输出的是一个VCF文件。 TNhaplotyper算法需要以下ALGO_OPTION: - `--tumor_sample SAMPLE_NAME`:BAM文件中肿瘤样本的SM标签名称。 根据运行模式,TNhaplotyper算法可能需要以下ALGO_OPTION: - `--normal_sample SAMPLE_NAME`:BAM文件中正常样本的SM标签名称。 - `--detect_pon`:表示您正在使用TNhaplotyper算法创建将成为正常样本面板一部分的VCF文件。 - `--cosmic COSMIC_VCF`:用于创建正常样本面板文件的体细胞突变目录(COSMIC)VCF文件的位置。仅支持单个文件。 - `--pon PANEL_OF_NORMAL_VCF`:包含在正常样本面板分析中检测到的变异的文件位置,用于去除假阳性。仅支持单个文件。 TNhaplotyper算法接受以下可选的ALGO_OPTION: - `--dbsnp dbSNP_FILE`:单核苷酸多态性数据库(dbSNP)的位置。dbSNP中的变异更有可能被标记为胚系变异,因为它们需要更多证据证明在正常样本中不存在。仅支持单个文件。 - `--min_base_qual QUALITY`:确定用于变异检测的碱基质量过滤。任何质量低于QUALITY的碱基将被忽略。默认值为10。 - `--prune_factor FACTOR`:局部组装中的最小剪枝因子;支持k-mer数量少于FACTOR的路径将从图中剪除。默认值为2。 - `--pcr_indel_model MODEL`:用于或多或少激进地剔除假阳性插入/缺失的PCR插入/缺失模型。可能的模式有: NONE(用于无PCR样本),HOSTILE、AGGRESSIVE和CONSERVATIVE,按激进程度递减排序。默认值为HOSTILE。 - `--phasing [1/0]`:在输出中启用或禁用的相位信息的标志。 - `--min_init_tumor_lod NUMBER`:初始通过检测变异时的最小肿瘤对数比值。默认值为4。 - `--min_init_normal_lod NUMBER`:初始通过检测变异时的最小正常对数比值。默认值为0.5。 - `--min_tumor_lod NUMBER`:最终变异检测时的最小肿瘤对数比值。默认值为6.3。 - `--min_normal_lod NUMBER`:用于检查肿瘤变异是否非正常变异的最小正常对数比值。默认值为2.2。 - `--min_strand_bias_lod NUMBER`:检测链偏好性的最小对数比值。默认值为2。 - `--min_strand_bias_power NUMBER`:检测链偏好性的最小效检力。默认值为0.9。 - `--min_pir_median NUMBER`:最小读段位置中位数。默认值为10。 - `--min_pir_mad NUMBER`:最小读段位置中位数绝对偏差。默认值为3。 - `--max_normal_alt_cnt NUMBER`:正常堆积中变异等位基因计数的最大值。默认值为2。 - `--max_normal_alt_qsum NUMBER`:正常堆积中变异等位基因质量得分总和的最大值。默认值为20。 - `--max_normal_alt_frac NUMBER`:正常堆积中变异等位基因的最大比例。默认值为0.03。 - `--tumor_contamination_frac NUMBER`:来自其他样本对肿瘤样本的污染比例估计值。默认值为0。 - `--normal_contamination_frac NUMBER`:来自其他样本对正常样本的污染比例估计值。默认值为0。 - `--filter_clustered_read_position`:过滤在测序读段开始或结束处聚集的变异。 - `--filter_strand_bias`:过滤显示链偏好性证据的变异。 - `--bam_output OUTPUT_BAM`:输出一个 BAM 文件,其中包含变异检测执行局部重组后修改过的读段。此选项应仅与较小的 BED 文件连用,用于故障排查目的。 - `--trim_soft_clip`:确定是否应从变异检测中排除读段中的软剪切碱基。 #### (25)TNhaplotyper2 TNhaplotyper2算法使用基于单倍型(Haplotype)的算法对肿瘤-正常配对样本或肿瘤和正常样本面板数据执行体细胞变异检测。 TNhaplotyper2算法的输入是一个或多个BAM文件;其输出是一个VCF文件,将用于在TNfilter中过滤结果; TNhaplotyper2还会输出一个与输出文件同名但扩展名为`.stats`的额外文件,其中包含有助于帮助过滤的统计信息。 TNhaplotyper2算法需要以下ALGO_OPTION: - `--tumor_sample SAMPLE_NAME`:BAM文件中肿瘤样本的SM标签名称。 根据运行模式,TNhaplotyper2算法可能需要以下ALGO_OPTION: - `--normal_sample SAMPLE_NAME`:BAM文件中正常样本的SM标签名称。 - `--pon PANEL_OF_NORMAL_VCF`:包含在正常样本面板分析中检测到的变异的文件位置,用于去除假阳性。仅支持单个文件。 TNhaplotyper2算法接受以下可选的ALGO_OPTION: - `--min_base_qual QUALITY`: 确定变异检测中使用的碱基质量过滤。任何质量低于QUALITY的碱基将被忽略。默认值为10。 - `--prune_factor FACTOR`:局部组装中的最小剪枝因子;支持k-mer数量少于FACTOR的路径将从图中剪除。将剪枝因子设置为0将启用自适应剪枝。默认值为0。 - `--pcr_indel_model MODEL`:用于或多或少激进地剔除假阳性插入/缺失的PCR插入/缺失模型。可能的模式有: NONE(用于无PCR样本),HOSTILE、AGGRESSIVE和CONSERVATIVE,按激进程度递减排序。默认值为CONSERVATIVE。 - `--min_init_tumor_lod NUMBER`:初始通过检测变异时的最小肿瘤对数比值。默认值为2.0。 - `--min_tumor_lod NUMBER`:最终变异检测时的最小肿瘤对数比值。默认值为3.0。 - `--min_normal_lod NUMBER`:用于检查肿瘤变异是否非正常变异的最小正常对数比值。默认值为2.2。 - `--germline_vcf VCF`:包含群体等位基因频率的VCF文件位置。 - `--default_af AF`:确定在胚系VCF中未找到的等位基因的等位基因频率值。在运行肿瘤-正常模式时默认值为1*10的负6次方,在不使用配对正常样本的仅肿瘤模式下运行时默认值为5 *10的负8次方。 - `--max_germline_af AF`:确定仅肿瘤模式下的最大胚系等位基因频率。默认值为0.01。 - `--call_pon_sites`:确定是否调用候选变异,即使它们存在于输入的正常样本对照集中。 - `--callable_depth DEPTH`: 确定一个位点被视为可检测的最小深度,用于包含可帮助过滤的统计信息的额外.stats文件。 - `--given GIVEN_VCF`:使用GIVEN_VCF中提供的变异执行变异检测。除了在发现模式下寻找变异外,还将评估文件中提供的位点和等位基因,但仅当变异的FILTER列为PASS或.时才进行。 - `--bam_output OUTPUT_BAM`:输出包含变异检测后局部重组修改读段的BAM文件。此选项应仅与小型bed文件一起用于故障排除的目的。 - `--trim_soft_clip`:确定是否应从变异检测中排除读段中的软剪切碱基。 - `--call_germline_sites`:确定是否检测在胚系位点中存在的候选变异。 #### (26)TNfilter TNfilter算法对TNhaplotyper2的输出执行过滤。 TNfilter算法的输入是一个包含待过滤候选变异的VCF文件;其输出是包含已过滤变异的VCF文件;TNfilter还会输出一个与输出文件同名但扩展名为.stats的额外文件,其中包含有关过滤的统计信息。 TNfilter算法需要以下ALGO_OPTION: - `-v CANDIDATE_VCF`:包含TNhaplotyper2生成的未过滤变异的文件位置和文件名。您可以使用经bgzip压缩并建立索引的VCF文件。除此该文件外,二进制文件还需要由TNhaplotyper2创建的名为CANDIDATE_VCF.stats的统计信息文件。 - `--tumor_sample SAMPLE_NAME`:BAM文件中肿瘤样本的SM标签名称。 根据运行模式的不同,TNfilter算法可能需要以下ALGO_OPTION: - `--normal_sample SAMPLE_NAME`:BAM文件中正常样本的SM标签名称。 TNfilter算法接受以下可选的ALGO_OPTION: - `--orientation_priors PRIORS`:包含OrientationBias生成的方向偏差信息的文件位置和文件名。此选项影响方向偏差过滤器。 - `--contamination FILE`:包含ContaminationModel生成的污染信息的文件位置和文件名。此选项影响污染过滤器。 - `--tumor_segments SEGMENTS`:包含ContaminationModel生成的肿瘤片段信息的文件位置和文件名。此选项影响胚系风险伪影过滤器。 - `--threshold_strategy STRATEGY`:确定应用于优化后验概率阈值的策略。可能的值有:f_score优先考虑F分数,precision优先考虑减少假阳性和constant。默认值为f_score。 - `--f_score_beta SCORE`:使用`--threshold_strategy f_score`时,确定过滤目标中召回率相对于精确度的权重。默认值为1,表示精确度和召回率权重相等。 - `--max_fp_rate FP_RATE`:使用`--threshold_strategy precision`时,确定预期的最大假阳性率。默认值为0.05。 - `--threshold THRESHOLD`:使用`--threshold_strategy constant`时,确定后验概率阈值。 - `--min_median_base_qual QUALITY`:确定变异 reads 的最小中值碱基质量。此选项会影响中值碱基质量过滤器。默认值为 20。 - `--max_event_count COUNT`:确定活跃区域允许的最大事件数。如果活跃区域中的事件超过COUNT,变异将被聚集事件过滤器过滤。默认值为2。 - `--unique_alt_reads COUNT`:确定需要支持变异的最小唯一ALT读段数。此选项会影响重复读段过滤器。默认值为0。 - `--max_mfrl_diff VALUE`:确定变异和参考片段中位数之间的最大差异。此选项会影响中位片段长度差异过滤器。默认值为10000。 - `--max_haplotype_distance DISTANCE`:确定同一单倍型内两个相位变异的最大距离。如果一个变异被过滤,DISTANCE内的所有相位变异也将被过滤掉,但超出DISTANCE的变异不会受到影响。此选项回影响单倍型过滤器。默认值为100。 - `--min_tumor_af AF`:确定最小肿瘤等位基因频率。此选项回影响低等位基因频率过滤器。默认值为0。 - `--min_median_map_qual QUALITY`:确定支持变异的读段的最小中位比对质量。此参数影响中位比对质量过滤器。默认值为30。 - `--long_indel_length LENGTH`:确定INDEL是否使用参考映射质量;长度超过LENGTH的INDEL将使用参考比对质量。此参数影响中位比对质量过滤器。默认值为5。 - `--max_alt_count COUNT`:确定一个位点的最大变异等位基因数。此选项会影响多等位基因过滤器。默认值为1。 - `--max_n_ratio RATIO`:确定N碱基与变异碱基的最大比率。此选项会影响N碱基比率过滤器。默认值为1。 - `--normal_p_value VALUE`::确定检测正常伪影的P值阈值。此选项会影响正常伪影过滤器。默认值为0.001。 - `--min_median_pos DISTANCE`:确定从变异到读段末端的最小中值距离。此选项会影响中值读段位置过滤器。默认值为1。 - `--min_slippage_length LENGTH`:确定STR中参考碱基的最小长度,超过此长度可能发生聚合酶滑移。此选项会影响聚合酶滑移过滤器。默认值为8。 - `--slippage_rate RATE`:确定可能发生聚合酶滑移区域的聚合酶滑移频率。此选项会影响聚合酶滑移过滤器。默认值为0.1。 - `--min_alt_reads_per_strand COUNT`:确定每条链上所需的最小变异读段数量。此选项影响硬链偏差过滤器。默认值为0。 除上述过滤器外,TNfilter还可能应用以下过滤器: - 基于模型的链偏差伪影过滤器,用于可能是链偏差伪影的位点。 - 正常样本面板过滤器,用于存在于正常样本面板中的位点。 - 肿瘤证据过滤器,用于变异证据较弱的位点。 #### (27)OrientationBias OrientationBias 算法用于估计测序数据中可能存在的任何方向偏差。 OrientationBias 算法的输入是一个或多个 BAM 文件;其输出的是一个包含方向偏差信息的文件,该文件将在 TNfilter 中过滤 TNhaplotyper2 的结果时使用。 OrientationBias 算法需要以下 ALGO_OPTION: - `--tumor_sample SAMPLE_NAME`:BAM 文件中肿瘤样本的 SM 标签名称。 OrientationBias 算法接受以下可选的 ALGO_OPTION: - `--min_base_qual QUALITY`:确定变异检测中使用的碱基的过滤质量。任何质量低于 QUALITY 的碱基将被忽略。默认值为 20。 - `--min_median_map_qual QUALITY`:决定最小中值比对质量。读段的中值比对质量低于 QUALITY 的位点将被忽略。默认值为 50。 - `--max_depth DEPTH`:确定分组的深度阈值。深度高于 DEPTH 的位点将被归为一组。默认值为 200。 #### (28)ContaminationModel ContaminationModel 算法用于估计跨样本污染和肿瘤分段,以便在 TNseq 流程中使用。 ContaminationModel 算法的输入是一个或多个 BAM 文件;其输出是一个包含污染信息的文件,该文件可在 TNfilter 中过滤 TNhaplotyper2 的结果时使用。此外,该工具可能会输出一个 .segments 文件,其中包含可帮助过滤的肿瘤分段信息。 ContaminationModel 算法需要以下 ALGO_OPTION: - `--tumor_sample SAMPLE_NAME`:BAM 文件中肿瘤样本的 SM 标签名称。 - `--vcf VCF`:包含群体等位基因频率的 VCF 文件位置。 根据运行模式的不同,ContaminationModel 算法可能需要以下 ALGO_OPTION: - `--normal_sample SAMPLE_NAME`:BAM 文件中正常样本的 SM 标签名称。 ContaminationModel 算法接受以下可选的 ALGO_OPTION: - `--tumor_segments CONTAMINATION.segments`:可选输出文件的位置和文件名,该文件包含肿瘤分段信息;此信息将在 TNfilter 中用于过滤可能是胚系风险伪影的调用。 - `--min_map_qual QUALITY`:确定使用的读段的过滤质量。任何比对质量低于 QUALITY 的读段将被过滤掉。默认值为 50。 - `--min_af AF`:确定群体等位基因频率的最小值。默认值为 0.01。 - `--max_af AF`:确定群体等位基因频率的最大值。默认值为 0.2。 #### (29)TNscope TNscope算法使用Haplotyper算法对肿瘤-正常配对样本或仅肿瘤数据执行体细胞变异检测。 TNscope算法的输入是BAM文件;其输出的是一个VCF文件。 TNscope算法需要以下ALGO_OPTION: - `--tumor_sample SAMPLE_NAME`:BAM文件中肿瘤样本的SM标签名称。 TNscope算法接受以下可选的ALGO_OPTION: - `--annotation 'ANNOTATION'`:确定将添加到输出VCF文件中的额外注释。目前支持FAD注释。在使用`--annotation FAD`选项时,将添加三个额外的注释:FAD、F1R2和F2R1。 - `--normal_sample SAMPLE_NAME`: BAM文件中正常样本的SM标签名称。在进行仅肿瘤体细胞变异检测时,不需要此参数。 - `--cosmic COSMIC_VCF`:用于创建正常样本面板文件的体细胞突变目录(COSMIC)VCF文件的位置。仅支持单个文件。 - `--pon PANEL_OF_NORMAL_VCF`:包含在正常样本面板分析中检测到的变异的文件位置,用于去除假阳性。仅支持单个文件。此文件与TNhaplotyper使用的文件相同。 - `--dbsnp dbSNP_FILE`:单核苷酸多态性数据库(dbSNP)的位置。dbSNP中的变异更可能被标记为胚系变异,因为它们需要更多证据证明在正常样本中不存在。仅支持单个文件。 - `--min_base_qual QUALITY`:确定变异检测中使用的碱基过滤质量。任何质量低于QUALITY的碱基将被忽略。默认值为15。 - `--prune_factor FACTOR`:局部组装中的最小剪枝因子;支持k-mer数量少于FACTOR的路径将从图中剪除。将剪枝因子设置为0将启用自适应修剪。默认值为2。 - `--pcr_indel_model MODEL`:用于或多或少激进地剔除假阳性插入/缺失的PCR插入/缺失模型。可能的模式有: NONE(用于无PCR样本),HOSTILE、AGGRESSIVE和CONSERVATIVE,按激进程度递减排序。默认值为CONSERVATIVE。 - `--phasing [1/0]`:在输出中启用或禁用的相位信息的标志。默认值为1(开启)。 - `--min_init_tumor_lod NUMBER`:初始通过检测变异时的最小肿瘤对数比值。默认值为4。 - `--min_init_normal_lod NUMBER`:初始通过检测变异时的最小正常对数比值。默认值为0.5。 - `--min_tumor_lod NUMBER`:最终变异检测时的最小肿瘤对数比值。默认值为6.3。 - `--min_normal_lod NUMBER`:用于检查肿瘤变异是否非正常变异的最小正常对数比值。默认值为2.2。 - `--min_dbsnp_normal_lod NUMBER`:在dbSNP位点检测正常非变异的最小对数比值。默认值为5.5。 - `--tumor_contamination_frac NUMBER`:来自其他样本的肿瘤样本污染比例估计。默认值为0。 - `--normal_contamination_frac NUMBER`:来自其他样本的正常样本污染比例估计。默认值为0。 - `--given GIVEN_VCF`:仅使用GIVEN_VCF中提供的变异执行变异检测。检测仅评估文件中提供的位点和等位基因,且仅当变异的FILTER列为PASS或.时才进行。 - `--bam_output OUTPUT_BAM`:输出包含变异检测后局部重组修改读段的BAM文件。此选项应仅与小型bed文件一起用于故障排除目的。 - `--disable_detector DETECTOR`:禁用特定检测器的变异检测:使用'sv'作为DETECTOR以防止检测结构变异,使用'snv_indel'作为DETECTOR以防止检测小变异。 - `--trim_soft_clip`:确定是否应从变异检测中排除读段中的软剪切碱基。 - `--trim_primer AMPLICON_TABLE`:确定是否根据AMPLICON_TABLE中提供的信息切除引物。AMPLICON_TABLE是一个 BED 文件,包含扩增子测序中引物的位置,作为一个包含八列的制表符分隔文件:`contig`, `amplicon_start`, `amplicon_end`, `name` (被忽略), `score` (被忽略), `strand` (被忽略), `insert_start`, `insert_end`;该工具将切除扩增子 reads 中位于 `amplicon_start` 与 `insert_start` 之间,以及 `amplicon_end` 与 `insert_end` 之间的碱基。 - `--trim_adaptor=[0|1]`:确定是否从输入读段中切除适配器碱基。默认值为切除适配器碱基。设置 `--trim_adaptor=0` 可将其关闭。 #### (30)RNASplitReadsAtJunction RNASplitReadsAtJunction算法通过去除N碱基但保留分组信息,并将任何悬挂进入内含子区域的序列进行硬切除,从而将读段拆分为外显子片段。 RNASplitReadsAtJunction算法的输入是BAM文件;其输出的也是BAM文件。 RNASplitReadsAtJunction算法需要以下ALGO_OPTION: - `--reassign_mapq IN_QUAL:OUT_QUAL`:该算法将把比对质量从 IN_QUAL重新分配为 OUT_QUAL。此参数是必需的,因为 STAR 将良好的比对质量分配为 255,而不是预期的默认分值 60。 RNASplitReadsAtJunction算法接受以下可选的ALGO_OPTION: - `--ignore_overhang`:确定是否忽略且不修复读段的悬挂部分。 - `--overhang_max_bases NUMBER`:确定硬切除悬挂部分允许的最大碱基数,如果悬挂部分的碱基数超过此值,则不会进行硬切除。默认值为40。 - `--overhang_max_mismatches NUMBER`:确定非硬切除悬挂部分允许的最大错配数,如果错配数太高,则会对整个悬挂部分进行硬切除。默认值为1。 - `--cram_write_options compressor=[gzip|bzip2|lzma|rans]`:CRAM输出压缩选项。如果未定义,默认为`compressor=gzip+rans`。 - `--cram_write_options version=[2.1|3.0]`:CRAM输出版本选项。如果未定义,默认为`version=3.0`。 #### (31)ContaminationAssessment ContaminationAssessment 算法评估样本 BAM 文件中存在的污染;此算法的输出可用作 TNseq 和 TNscope 工具中 `contamination_frac`、`normal_contamination_frac` 和 `tumor_contamination_frac` 参数的值。 ContaminationAssessment 算法的输入是一个 BAM 文件;其输出是一个文本文件。 ContaminationAssessment 算法需要以下 ALGO_OPTION: - `--pop_vcf VCF_FILE`:包含样本特定人群等位基因频率信息的 VCF 文件位置。 - `--genotype_vcf VCF_FILE`:包含为个体报告的 DNAseq 变异的 VCF 文件位置;要计算肿瘤样本中的污染,应使用为正常样本报告的 DNAseq 变异。您可以通过在样本BAM上使用 Haplotyper 或 Genotyper 来创建此文件。 ContaminationAssessment 算法接受以下可选的 ALGO_OPTION: - `--type ASSESS_TYPE`:确定估计的类型。可能的值为 SAMPLE、READGROUP 和 META,分别用于按样本、按测序通道或跨所有读段进行污染评估。允许包含该选项多个实例,以在多个层面评估污染。默认值为 META。 - `--min_base_qual QUALITY`:确定污染评估中使用的碱基的过滤质量。任何质量低于 QUALITY 的碱基将被忽略。默认值为 20。 - `--min_map_qual QUALITY`:确定污染评估中使用的读段的过滤质量。任何质量低于 QUALITY 的读段将被忽略。默认值为 20。 - `--min_basecount NUMBER`:确定在评估污染之前在一个位点需要存在的最小碱基数。默认值为 500。 - `--trim_thresh NUMBER`:用于修剪位点的阈值;如果污染比率大于 0.5 的概率大于阈值,则该位点不会包含在污染评估中。默认值为 0.95。 - `--trim_frac NUMBER`:确定基于修剪阈值可能被修剪的最大位点比例。默认值为 0.01。 - `--precision NUMBER`:确定输出百分比数字的精度。默认值为 0.1。 - `--base_report FILE`:包含处理数据扩展报告的输出文件的位置和文件名。 - `--population POPULATION_NAME`:用于确定样本基线等位基因频率的人群。默认值为 CEU。 #### (32)TNModelApply TNModelApply 算法将机器学习模型应用于 TNscope 的结果,以帮助进行变异过滤。此算法仅在 Sentieon Genomics 软件的 Linux 版本中受支持。 TNModelApply 算法在driver级别没有输入,其输出是一个 VCF 文件。 TNModelApply 算法需要以下 ALGO_OPTION: - `-v INPUT`:来自 TNscope 变异检测输出的 VCF 文件位置;您可以使用经 bgzip 压缩并建立索引的 VCF 文件。 - `-m MODEL_FILE`:包含机器学习模型的文件位置。 TNModelApply 算法通过向变异添加 MLrejected FILTER 来修改输入的 VCF 文件;由于添加了 FILTER,您可能希望删除输入 VCF 中已存在的任何 FILTER,因为它们可能不再相关。您可以使用 bcftools(https://samtools.github.io/bcftools/bcftools.html )来实现这一目的: ``` $BCF/bcftools annotate -x "^FILTER/MLrejected,FILTER/PASS" -O z \ -o $OUTPUT.vcf.gz $INPUT.vcf.gz ``` #### (33)DNAscope DNAscope算法执行改进版的Haplotype变异检测。 DNAscope算法的输入是BAM文件;输出的是一个VCF文件。 DNAscope算法接受以下可选的ALGO_OPTION: - `--annotation 'ANNOTATION_LIST'`:确定将添加到输出VCF的其他注释。使用逗号分隔的列表来启用或禁用注释。包括'none'以删除默认注释;在注释前加感叹号(!)以禁用特定注释。有关支持的注释,请参见 *第6.2节*。 - `-d dbSNP_FILE`:用于标记已知变异的单核苷酸多态性数据库(dbSNP)的位置。仅支持单个文件。 - `--var_type VARIANT_TYPE`:确定将检测哪些变异类型;VARIANT_TYPE是以下可能值的逗号分隔列表: - SNP :检测单核苷酸多态性。这包含在默认行为中。 - INDEL :检测插入-缺失。这包含在默认行为中。 - BND :检测结构变异检测器所需的断点信息。 - `--call_conf CONFIDENCE`:确定检测变异的变异质量阈值。质量低于CONFIDENCE的变异将被移除。 - `--emit_conf CONFIDENCE`:确定输出变异的变异质量阈值。质量低于CONFIDENCE的变异将不会添加到输出VCF文件中。 - `--emit_mode MODE`:确定将输出哪些检测结果。MODE的可能值为: - variant:仅在置信的变异位点输出检测结果。这是默认行为。 - confident:在置信的变异位点或置信的参考位点输出检测结果。 - all:输出所有检测结果,无论其置信度如何。 - gvcf:输出联合检测所需的额外信息。如果要使用GVCFtyper算法执行联合检测,则需要此选项。 - `--gq_bands LIST_OF_BANDS`:确定用于压缩具有相似基因型质量(GQ)的变异的波段,这些变异将在GVCF输出文件中作为单个VCF记录输出。LIST_OF_BANDS是一个逗号分隔的波段列表,其中每个波段由START-END/STEP定义。默认值为1-60,60-99/10,99。 - `--min_base_qual QUALITY`:确定变异检测中使用的碱基的过滤质量。任何质量低于QUALITY的碱基将被忽略。默认值为10。 - `--pcr_indel_model MODEL`:用于或多或少激进地剔除假阳性插入缺失的PCR插入缺失模型。可能的MODEL有:NONE(用于无PCR样本),以及HOSTILE、AGGRESSIVE和CONSERVATIVE,按激进程度递减排序。默认值为CONSERVATIVE。 - `--phasing [1/0]`:在输出中启用或禁用的相位的标志。默认值为1(开启)。相位仅针对二倍体样本进行计算。 - `--ploidy PLOIDY`:确定正在处理的样本的倍性数。默认值为2。 - `--prune_factor FACTOR`:局部组装中的最小剪枝因子;支持k-mer数量少于FACTOR的路径将从图中剪除。默认值为2。 - `--trim_soft_clip`:确定是否应从变异检测中排除读段中的软剪切碱基。仅建议处理RNA读段时使用此参数。 - `--given GIVEN_VCF`:仅使用GIVEN_VCF中提供的变异执行变异检测。检测将仅评估文件中提供的位点和等位基因,并且仅当变异的FILTER列为PASS或.时才进行。此选项不能与`--emit_mode gvcf`一起使用。 - `--bam_output OUTPUT_BAM`:输出包含变异检测所做的局部重组后修改读段的BAM文件。此选项仅应与小型bed文件一起用于故障排除目的。 - `--filter_chimeric_reads`:确定在检测变异时是否使用嵌合读段。默认情况下,仅当设置了`var_type BND`时才包括嵌合读段。 - `--trim_adaptor`:确定是否从输入读段中切除接头碱基。默认值为不切除接头碱基,因为那可能会影响结构变异周围的区域。 - `--model MODEL_FILE`:将与DNAModelApply工具一起使用的机器学习模型文件的位置;该模型将用于确定变异检测中使用的设置。 #### (34)DNAModelApply DNAModelApply 算法执行使用 DNAscope 进行变异调用的第二个步骤。此算法仅在 Sentieon Genomics 软件的 Linux 版本中支持。 DNAModelApply 算法在driver级别没有输入,其输出是一个包含变异的 VCF 文件。 DNAModelApply 算法需要以下 ALGO_OPTION: - `-v INPUT`:来自 DNAscope 变异检测算法的 VCF 文件位置,该算法需输入模型文件以确定正确的设置执行。您可以使用经 bgzip 压缩并建立索引的 VCF 文件。 - `-m MODEL_FILE`:机器学习模型文件的位置;此文件应与 DNAscope 命令中用于生成输入 VCF 的文件相同。 DNAModelApply 算法不支持任何可选的 ALGO_OPTION。 DNAModelApply 算法通过向变异添加 MLrejected FILTER 来修改输入的 VCF 文件;由于添加了 FILTER,您可能希望删除输入 VCF 中已存在的任何 FILTER,因为它们可能不再相关。您可以使用 bcftools(https://samtools.github.io/bcftools/bcftools.html )来实现这一目的: ``` $BCF/bcftools annotate -x "^FILTER/MLrejected,FILTER/PASS" -O z \ -o $OUTPUT.vcf.gz $INPUT.vcf.gz ``` #### (35)SVSolver SVSolver 算法执行样本的结构变异检测,前提是该样本之前已使用 DNAscope 算法进行处理,并使用了` --var_type bnd` 选项。 SVSolver 算法在driver级别没有输入,其输出的是一个包含检测到结构变异的 VCF 文件。 SVSolver 算法需要以下 ALGO_OPTION: - `-v INPUT`: 使用 `--var_type bnd` 选项对样本执行 DNAscope 变异检测算法所得到的 VCF 文件的位置。您可以使用 bgzip 压缩并建立索引的 VCF 文件。 SVSolver 算法不支持任何可选的 ALGO_OPTION。 #### (36)VariantPhaser VariantPhaser 算法通过使用包含长读段的 BAM 文件中的读段信息,对 VCF 文件中的变异进行基于读段的相位处理。 VariantPhaser 算法的输入是一个 BAM 文件和一个 VCF 文件;其输出是相位处理后的 VCF 文件。 VariantPhaser 算法需要以下 ALGO_OPTION: - `-v INPUT`:包含需要进行相位处理的变异的 VCF 文件的位置。您可以使用 bgzip 压缩并建立索引的 VCF 文件。 ### 5.1.3 DRIVER read_filter 选项 DRIVER二进制程序的`--read_filter`参数允许在执行计算之前过滤或转换输入BAM文件中的读段。可以在同一命令行中使用多个`--read_filter`参数,它们将按顺序执行,因此顺序很重要。 参数语法为:`--read_filter FILTER,OPTION=VALUE,OPTION=VALUE,...` #### (1)QualCalFilter read_filter QualCalFilter read过滤器用于转换读段并执行碱基质量分数重校准,同时修改重校准表中包含的信息。 QualCalFilter read过滤器需要以下选项之一: - `table=TABLE_FILEPATH`:用作执行碱基质量分数重校准基础的重校准表的位置。 - `use_oq=[true/false]`:确定是否使用BAM文件中OQ标签包含的原始碱基质量分数。此选项不能与table选项一起使用,它用于通过将输出的碱基质量分数设置为输入BAM文件中OQ标签中包含的分数来撤销碱基质量分数重校准。通常,此选项将在第二个QualCalFilter read过滤器之前使用,以首先撤销输入BAM文件上可能进行的重校准。 QualCalFilter read过滤器接受以下可选选项: - `prior=PRIOR`:确定所有碱基质量分数的全局偏差。 - `min_qual=QUAL`:确定执行重校准的质量阈值;质量分数小于QUAL的碱基将不会被重校准。 - `levels=LEVEL1/LEVEL2/...`:确定碱基质量分数的静态量化级别。 - `indel=[true/false]`:确定是否将INDEL的碱基质量分数添加到BAM标签中。 - `keep_oq=[true/false]`:确定是否通过使用BAM文件中的OQ标签来保留重校准前的原始碱基质量分数。 #### (2)OverclippingFilter read_filter OverclippingFilter read过滤器用于根据读段的软剪切特征对其进行过滤。 OverclippingFilter read过滤器接受以下可选选项: - `min_align_len=LENGTH`:过滤非软剪切碱基数少于LENGTH的读段。 - `count_both_ends=[true/false]`:如果设置为true,则仅过滤读段两端都有软剪切的读段,这样只有一端有软剪切的读段将不会被过滤,无论其非软剪切长度如何。默认值为true。 #### (3)SimplifyCigarTransform read_filter SimplifyCigarTransform read过滤器用于按以下规则简化和标准化CIGAR: - 长度为零的操作符:删除。 - EQUAL操作符(=)转换为MATCH操作符(M)。 - DIFF操作符(X)转换为MATCH操作符(M)。 - 相邻的INS(I)和DEL(D)操作符合并为MATCH(M)。 - 尾部DEL操作符(D):删除。 ## 5.2 BWA 二进制程序 BWA二进制程序用于执行DNA-seq数据的比对。 在Sentieon®202112版本中,修复了原始bwa mem中导致少量比对的MAPQ不正确的bug。提供了一个兼容性环境变量`ksw_compat=1`,设置此环境变量将使Sentieon® bwa mem的行为与http://bio-bwa.sourceforge.net/bwa.shtml 中描述的相同,为某些比对提供不正确的MAPQ。默认行为为所有比对实现了正确的MAPQ计算,并在一些较新的硬件上提供了速度提升。 BWA二进制程序有两种重要模式:"mem"模式用于将FASTQ文件与参考FASTA文件进行比对,以及"shm"模式用于将FASTA索引文件加载到内存中,以便在同一服务器上运行的多个BWA进程之间共享。 ### 5.2.1 BWA mem语法 您可以运行以下命令将单端FASTQ1文件或一对双端FASTQ文件与FASTA参考序列进行比对,比对后的 reads 将产生到标准输出,以便通过管道传输到util sort: ``` <SENTIEON_FOLDER>/bin/sentieon bwa mem OPTIONS FASTA FASTQ1 [FASTQ2] ``` 此命令的参数(OPTIONS)包括: - `-t NUMBER_THREADS`:软件将用于运行并行进程的计算线程数。此参数是可选的;如果省略,bwa二进制工具将使用1个线程。 - `-p`:确定第一个输入FASTQ文件是否包含交错的双端读段。如果使用此参数,则仅使用单个FASTQ输入,因为第二个FASTQ2文件将被忽略。 - `-M`:确定是否将分裂读段标记为次要。 - `-R READGROUP_STRING`:所有读段将附加的读组头行。推荐的是@RG\tID:$readgroup\tSM:$sample\tPL:$platform\tPU:$platform_unit - `$readgroup`是识别读段的唯一 ID。 - `$sample`是读段所属的样本的名称。 - `$platform`是测序技术,通常是ILLUMINA。 - `$platform_unit`是执行测序的测序单位。 - `-K CHUNK_SIZE`:确定将同时比对的读段组的大小。如果未设置此参数,结果将取决于使用的线程数。 ### 5.2.2 BWA shm语法 您可以运行以下命令将FASTA索引文件加载到内存中: ``` <SENTIEON_FOLDER>/bin/sentieon bwa shm FASTA ``` 您可以运行以下命令列出存储在内存中的FASTA索引文件: ``` <SENTIEON_FOLDER>/bin/sentieon bwa shm -l ``` 您可以运行以下命令删除存储在内存中的所有FASTA索引文件,从而在不再需要时释放内存: ``` <SENTIEON_FOLDER>/bin/sentieon bwa shm -d ``` 此命令的参数(OPTIONS)包括: - `-t NUMBER_THREADS`:软件将用于运行并行进程的计算线程数。此参数是可选的;如果省略,bwa二进制工具将使用1个线程。 - `-f FILE`:将用于减少峰值内存使用的临时文件的位置。 ### 5.2.3 控制BWA中的内存使用 默认情况下,BWA在Linux系统中将使用约24 GB,在Mac系统中将使用8 GB。您可以通过bwt_max_mem环境变量控制内存使用,这可以通过使用更多内存来提高速度性能,或者以牺牲速度性能为代价限制内存使用。例如,通过在脚本中添加以下内容,您将获得更快的比对: ``` export bwt_max_mem=50G ``` 请记住,您在bwt_max_mem环境变量中使用的数字不是硬限制,而是BWA使用内存的估计值;因此,如果BWA内存使用不超过某个特定值以下的任何值,这意味着它是特定参考所需的最小内存,设置为小于最小所需内存的值不会改变BWA mem作业的内存使用。 ### 5.2.4 使用现有BAM文件作为输入 如果您无法访问FASTQ输入,而只有已比对和排序的BAM文件,您可以通过运行samtools将其用作输入并重新进行比对: ``` samtools collate -@ 32 -Ou INPUT_BAM tmp- | samtools fastq -@ 32 -s \ /dev/null -0 /dev/null - | <SENTIEON_FOLDER>/bin/sentieon bwa mem -t 32 -R \ '@RG\tID:id\tLB:lib\tSM:sample\tPL:ILLUMINA' -M -K 1000000 -p $ref /dev/stdin \ | <SENTIEON_FOLDER>/bin/sentieon util sort -t 32 -o OUTPUT_BAM --sam2bam - ``` 或者,您可以先创建FASTQ文件,然后像通常那样处理它们: ``` samtools collate -n -@ 32 -uO INPUT_BAM tmp- | samtools fastq -@ 32 \ -s >(gzip -c > single.fastq.gz) -0 >(gzip -c > unpaired.fastq.gz) \ -1 >(gzip -c > output_1.fastq.gz) -2 >(gzip -c > output_2.fastq.gz) - ``` 如果您这样做,可能会遇到BWA中异常的内存使用;如果出现这种情况,您可以按照 *第7.2.5 使用从 BAM 文件创建的 FASTQ 文件时使用异常数量的内存* 中的说明进行操作。 ## 5.3 STAR 二进制程序 STAR 二进制程序用于执行 RNA-seq 数据的比对,其行为与https://github.com/alexdobin/STAR 中描述的工具相同。 ### 5.3.1 使用压缩的 FASTQ.gz 输入文件 使用压缩的 FASTQ 输入文件需要使用 STAR 选项 --readFilesCommand COMMAND 来确定将使用哪个外部程序来解压输入文件。当使用 Sentieon® 实现的 STAR 与 Sentieon® util sort 一起使用时,高效的输入处理可能会导致解压成为瓶颈,因此使用快速解压方法非常重要。 ## 5.4 Minimap2 二进制程序 minimap2 二进制程序用于执行 PacBio 或 Oxford Nanopore 基因组测序数据的比对,其行为与 https://github.com/lh3/minimap2 中描述的工具相同。 ## 5.5 UTIL 二进制程序 UTIL是用于运行一些实用功能的二进制程序。这个二进制程序主要用于处理来自BWA的原始读段输出。 ### 5.5.1 UTIL 语法 UTIL二进制程序的一般语法是: ``` sentieon util MODE [OPTIONS] ``` 该命令支持的模式(MODE)包括: - `index`:为BAM文件建立索引。以下命令将在输入文件相同位置生成bai BAM索引文件: ``` sentieon util index INPUT.bam ``` - `vcfindex`:为VCF文件建立索引。以下命令将在输入文件相同位置生成idx VCF索引文件: ``` sentieon util vcfindex INPUT.vcf ``` - `sort`:对BAM文件进行排序。使用sort模式的UTIL命令的可选参数包括: - `-t NUMBER_THREADS`:软件用于运行并行进程的计算线程数。默认使用服务器上所有可用的线程。 - `-r REFERENCE`:参考FASTA文件的位置。如果使用CRAM输出文件则此参数是必需的,否则为可选。 - `-i INPUT`:输入文件的位置。 - `-o OUTPUT`:输出文件的位置和文件名。 - `--temp_dir DIRECTORY`:确定临时文件的存储位置。默认为运行命令的文件夹($PWD)。 - `--cram_read_options decode_md=0`:CRAM输入选项,用于关闭输入CRAM中的NM/MD标签。 - `--cram_write_options compressor=[gzip|bzip2|lzma|rans]`:CRAM输出压缩选项。如果未定义,默认为`compressor=gzip+rans`。 - `--cram_write_options version=[3.0|3.1]`:CRAM输出版本选项。如果未定义,默认为`version=3.0`。 - `--bam_compression COMPRESSION_LEVEL[0-9]`:输出BAM文件的gzip压缩级别。默认值为6。 - `--sam2bam`:表示输入将是未压缩的SAM文件,需要转换为BAM。如果不使用此选项,输入应该已经使用samtools从BWA输出转换为BAM格式。 - `--block_size BLOCK_SIZE`:用于排序的块大小。 - `--trim_primer AMPLICON_TABLE,clip=CLIPPING`:确定是否将引物标记为软/硬剪切,取决于CLIPPING的值是soft还是hard。当使用选项`--trim_primer AMPLICON_TABLE,clip=soft`时,需要确保在后续的变异调用命令中使用`--trim_soft_clip`选项,以便忽略软剪切的引物。引物基于AMPLICON_TABLE中提供的信息确定。AMPLICON_TABLE是一个BED文件,包含扩增子测序中引物位置的制表符分隔文件,有八列:contig、amplicon_start、amplicon_end、name(忽略)、score(忽略)、strand(忽略)、insert_start、insert_end;该工具将修剪amplicon_start和insert_start之间,以及amplicon_end和insert_end之间的扩增子读段碱基。 ``` sentieon util sort -t NUMBER_THREADS --sam2bam -i INPUT.sam -o OUTPUT.bam ``` - `vcfconvert`:压缩和解压VCF和GVCF文件。 以下命令将压缩并为输入文件建立索引 ``` sentieon util vcfconvert INPUT.vcf OUTPUT.vcf.gz ``` 以下命令将解压使用gzip生成的非索引vcf文件,然后压缩并建立索引。使用此命令时确保输入和输出文件不相同: ``` sentieon util vcfconvert INPUT.gz OUTPUT.vcf.gz ``` - `stream`:在流模式下执行碱基质量校正。使用stream模式的UTIL命令的可选参数包括: - `-r REFERENCE`:参考FASTA文件的位置。如果使用CRAM输出文件则此参数是必需的,否则为可选。 - `-i INPUT`:输入文件的位置。默认情况下二进制程序将使用stdin作为输入。 - `-q RECAL_TABLE`:重校准表的位置。 - `-t NUMBER_THREADS`:软件用于运行并行进程的计算线程数。默认值为1。 - `-o OUTPUT`:输出文件的位置和文件名。默认情况下二进制程序将输出到stdout,以便能够流式传输结果。 - `--output_format FORMAT`:确定输出流的格式。可能的FORMAT值为BAM或CRAM。默认为BAM。 - `--output_index_file OUTPUT_INDEX`:确定将在何处创建相应的索引文件。如果省略此选项,工具将不生成索引文件。 - `--read_filter FILTER,OPTION=VALUE,OPTION=VALUE`:在应用算法之前执行读段过滤或转换。请参阅driver使用说明的 *第5.1.3节* 获取有关可用过滤器及其功能的其他信息。 - `--cram_read_options decode_md=0`:CRAM输入选项,用于关闭输入CRAM中的NM/MD标签。 - `--cram_write_options compressor=[gzip|bzip2|lzma|rans]`:CRAM输出压缩选项。如果未定义,默认为compressor=gzip。 - `--cram_write_options version=[3.0|3.1]`:CRAM输出版本选项。如果未定义,默认为`version=3.0`。 - `--bam_compression COMPRESSION_LEVEL[0-9]`:输出BAM文件的gzip压缩级别。默认值为6。 以下命令将应用重校准并将相应的重校准BAM文件输出到stdout: ``` sentieon util stream -i INPUT.bam -q RECAL_TABLE \ --output_index_file OUTPUT.bam.bai -o - ``` 除非包含`output_index_file`选项,否则上述命令不会生成索引文件。 ## 5.6 UMI 二进制程序 UMI是用于处理包含UMI序列的读段的二进制程序。 ### 5.6.1 UMI 语法 UMI二进制程序的一般语法是: ``` sentieon umi MODE [OPTIONS] ``` 该命令支持的模式(MODE)包括: - `extract`:预处理包含UMI序列读段的FASTQ文件。extract模式的语法是: ``` sentieon umi extract [OPTIONS] read_structure fastq1 [fastq2] [fastq3] ``` 其中: - `read_structure`是读段的逻辑结构。它由一系列“整数+字符”对组成,描述了“碱基数量+类型”;类型可以是M(分子条形码/Molecular barcode)、T(模板/Template)和 S(跳过/Skip)。读结构由逗号分隔的组组成,每组将从相应的输入 FASTQ 中读取(第一组来自第一个 FASTQ,第二组来自第二个 FASTQ,依此类推)。 - `fastq1/2/3`是FASTQ文件。最多支持3个输入FASTQ文件,以允许UMI序列已经在单独的FASTQ文件中的用例。 使用extract模式的UMI二进制程序的可选参数包括: - `-o OUTPUT`:输出文件的位置和文件名。如果省略,输出将是stdout。 - `-d`:如果存在,将以双端模式进行提取。 - `--umi_tag TAG`:逻辑UMI标签。默认值为XR。 ## 5.7 PLOT 脚本 PLOT是用于创建度量和重校准阶段结果图的脚本。图表存储在PDF文件中。 ### 5.7.1 PLOT 语法 PLOT脚本的一般语法是: ``` sentieon plot STAGE -o OUTPUPT_FILE INPUTS [OPTIONS] ``` 该命令支持的模式包括: - GCBias:从GCBias的度量结果生成PDF文件。 - QualDistribution:从QualDistribution的指标结果生成PDF文件。 - InsertSizeMetricAlgo:从InsertSizeMetricAlgo的指标结果生成PDF文件。 - MeanQualityByCycle:从MeanQualityByCycle的指标结果生成PDF文件。 - QualCal:从BQSR QualCal工具生成PDF文件。 - VarCal:从VQSR VarCal工具生成PDF文件。 #### (1) PLOT 绘制GCBias阶段的结果 生成GCBias阶段图的INPUTS是: - GC_METRIC_TXT:即 *第5.1.2节* 中GCBias算法的输出文件。 GCBias度量STAGE的绘图不接受任何参数。 #### (2)绘制MeanQualityByCycle阶段的结果 生成MeanQualityByCycle阶段图的INPUTS是: - MQ_METRIC_TXT:即 *第5.1.2节* 中MeanQualityByCycle算法的输出文件。 MeanQualityByCycle度量STAGE的绘图不接受任何参数。 #### (3)绘制QualDistribution阶段的结果 生成QualDistribution阶段图的INPUTS是: - QD_METRIC_TXT:即 *第5.1.2节* 中QualDistribution算法的输出文件。 QualDistribution度量STAGE的绘图不接受任何参数。 #### (4)绘制InsertSizeMetricAlgo阶段的结果 生成InsertSizeMetricAlgo阶段图的INPUTS是: - IS_METRIC_TXT:即 第5.1.2节 中InsertSizeMetricAlgo算法的输出文件。 InsertSizeMetricAlgo度量STAGE的绘图不接受任何参数。 #### (5)绘制QualCal阶段的结果 生成bqsr阶段图的INPUTS是: - RECAL_RESULT.CSV:即 *第5.1.2节* 中QualCal算法的输出csv文件。 bqsr STAGE的绘图不接受任何参数。 #### (6)脚本绘制VarCal阶段的结果 生成vqsr阶段图的INPUTS是: - PLOT_FILE:由 *第5.1.2节* 中VarCal算法创建的文件,包含创建报告所需的数据。 vqsr STAGE的绘图接受以下OPTIONS: - `tranches_file=TRANCHES_FILE`:包含调用集分区到质量梯度的文件位置,由 *第5.1.2节* 中的VarCal算法生成。 - `target_titv=TITV_THRES`:物种的预期TiTv(转换/颠换)比例;用于计算图中的真阳性和假阳性数量。 - `min_fp_rate=MIN_RATE`:最小假阳性数;用于计算图中的真阳性和假阳性数量。 ## 5.8 LICSRVR二进制程序 LICSRVR是用于运行授权许可服务器的二进制程序,用于促进动态授权许可分配和记录集群内的授权许可使用情况。 ### 5.8.1 LICSRVR语法 LICSRVR二进制程序的一般语法是: ``` <SENTIEON_FOLDER>/bin/sentieon licsrvr [--start|--stop] [--log LOG_FILE] LICENSE_FILE ``` 该命令的以下输入是可选的: - `LOG_FILE`:包含服务器日志的输出文件的位置和文件名。 - `LICENSE_FILE`:服务器授权许可文件的位置。 授权许可服务器运行后,客户端应用程序可以通过将SENTIEON_LICENSE环境变量设置为形式为HOST:PORT的服务器地址来从服务器请求授权许可令牌。 licsrvr二进制程序支持以下附加模式: - `--version`:将报告二进制程序所属的软件包版本。 - `--dump`:将报告许可证服务器的当前状态,包括可用许可证的数量。 ``` <SENTIEON_FOLDER>/bin/sentieon licsrvr --dump LICENSE_FILE ``` - `--dump=update`:如果授权许可信息已更新并被授权许可服务器自动拉取,此模式将更新的许可证信息转储到stdout。以下命令将报告授权许可服务器拥有的更新授权许可信息(如果有任何更改): ``` <SENTIEON_FOLDER>/bin/sentieon licsrvr --dump=update LICENSE_FILE ``` ## 5.9 LICCLNT二进制程序 LICCLNT是用于测试许可证服务器功能的二进制程序,帮助确定授权许可服务器是否运行,以及不同算法有多少可用授权许可。 ### 5.9.1 LICCLNT语法 LICCLNT二进制程序有两种模式,一种用于ping授权许可服务器,另一种用于检查特定算法的可用授权许可。 你可以运行以下命令来检查授权许可服务器是否运行: ``` <SENTIEON_FOLDER>/bin/sentieon licclnt ping --server HOST:PORT ``` 如果服务器运行,命令将返回0。 你可以运行以下命令来检查有多少授权许可: ``` <SENTIEON_FOLDER>/bin/sentieon licclnt query --server HOST:PORT FEATURE ``` 该命令将返回特定授权许可功能可用的授权许可数量,这可用于管理你的作业:在提交使用特定线程数的作业之前,你可以检查是否有足够的授权许可用于这些线程,防止工具在等待许可证时闲置。 *** # 6 工具功能 ## 6.1 处理多个输入文件 通常,有两种情况需要同时处理多个输入文件:单个样本在多个测序通道中测序,或者处理来自一个群体的多个样本。 ### 6.1.1 处理来自同一样本的多个输入文件 当您有多于一组输入 fastq 文件时,应该为每组输入 fastq 文件执行单独的比对阶段,然后使用多个排序后的 BAM 文件作为下一阶段的输入。下一阶段将负责将所有 BAM 文件合并为一个。您需要确保每组输入 fastq 文件使用相同的 SM 样本名称但不同的 RG 读组名称进行比对,以便后续阶段能正确处理不同的数据。 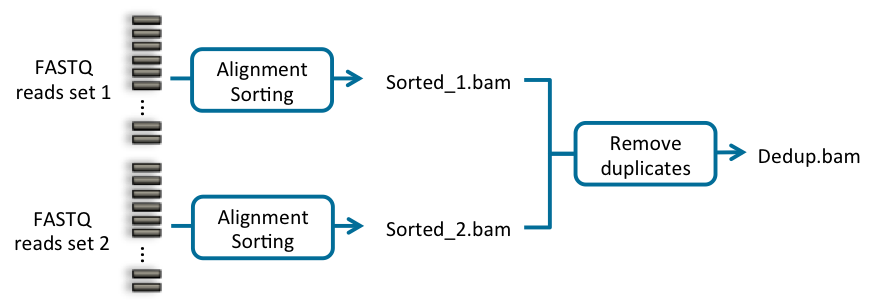 <center>图6-1 使用来自同一样本的多个输入文件的生物信息学流程</center> ``` #Run alignment for 1st input file set (sentieon bwa mem -R '@RG\tID:GROUP_NAME_1 \tSM:SAMPLE_NAME \tPL:PLATFORM' \ -t NUMBER_THREADS REFERENCE SAMPLE_1 [SAMPLE_1_2] || echo -n 'error' ) \ | sentieon util sort -o SORTED_BAM_1 -t NUMBER_THREADS --sam2bam -i - #Run alignment for 2nd input file set (sentieon bwa mem -R '@RG\tID:GROUP_NAME_2 \tSM:SAMPLE_NAME \tPL:PLATFORM' \ -t NUMBER_THREADS REFERENCE SAMPLE_2 [SAMPLE_2_2] || echo -n 'error' \ sentieon util sort -o SORTED_BAM_2 -t NUMBER_THREADS --sam2bam -i - #Run dedup on both BAM files sentieon driver -t NUMBER_THREADS -i SORTED_BAM_1 -i SORTED_BAM_2 \ --algo LocusCollector --fun score_info SCORE_TXT sentieon driver -t NUMBER_THREADS -i SORTED_BAM_1 -i SORTED_BAM_2 \ --algo Dedup --rmdup --score_info SCORE_TXT --metrics DEDUP_METRIC_TXT DEDUPED_BAM ``` 另外,您也可以将多个 BWA 比对的结果输入到单个 util sort 命令中,以尽快将结果合并到一个排序好的 bam 文件中。您仍然需要确保每组输入的 fastq 文件在比对时使用相同的 SM 样本名称,但使用不同的 RG 读组名称,这样后续阶段才能正确处理不同的数据。 ``` #Run alignment for both input file sets sequentially (sentieon bwa mem -R '@RG\tID:GROUP_NAME_1\tSM:SAMPLE_NAME\tPL:PLATFORM' \ -t NUMBER_THREADS REFERENCE SAMPLE_1 [SAMPLE_1_2] && sentieon bwa mem -R \ '@RG\tID:GROUP_NAME_2 \tSM:SAMPLE_NAME \tPL:PLATFORM' \ -t NUMBER_THREADS REFERENCE SAMPLE_2 [SAMPLE_2_2] || echo -n 'error' ) \ | sentieon util sort -o SORTED_COMBINED_BAM -t NUMBER_THREADS --sam2bam -i -#Run dedup on combined BAM file sentieon driver -t NUMBER_THREADS -i SORTED_COMBINED_BAM_1 \ --algo LocusCollector --fun score_info SCORE_TXT sentieon driver -t NUMBER_THREADS -i SORTED_COMBINED_BAM_1 \ --algo Dedup --rmdup --score_info SCORE_TXT --metrics DEDUP_METRIC_TXT DEDUPED_BAM ``` ### 6.1.2 处理来自不同样本的多个输入文件 共同处理来自一个群体的多个样本(也称为联合变异检测)可以通过两种方式完成: - 单独处理每个样本,并使用 Haplotyper 算法的 `--emit_mode gvcf `选项创建包含额外信息的 GVCF 文件,然后使用 GVCFtyper 算法处理所有 GVCF。这种方法允许在处理额外样本后轻松快速地重新处理。 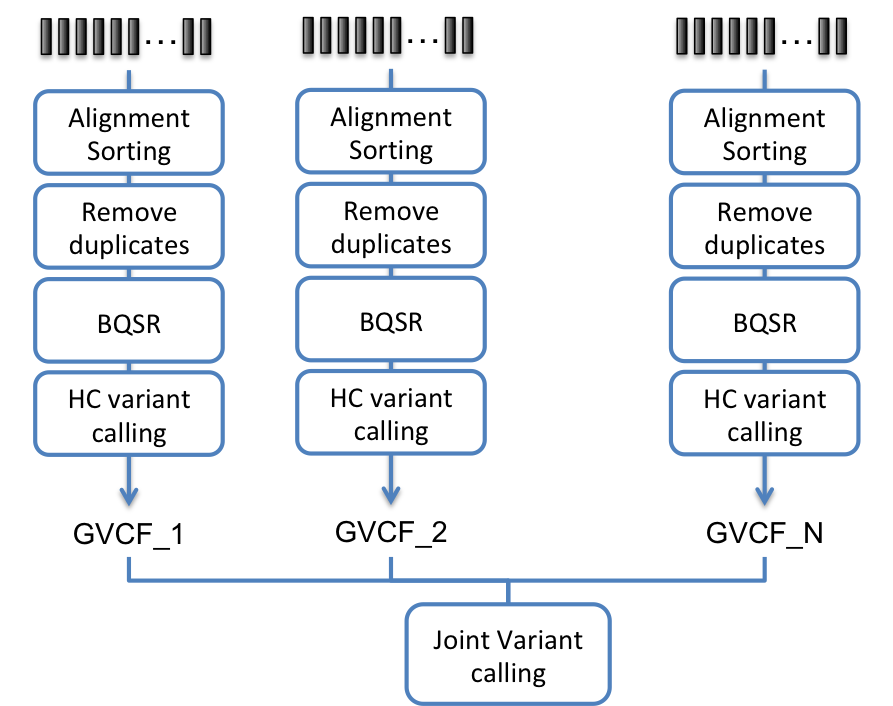 <center>图6-2 使用 GVCF 输出模式的 Haplotyper 和 GVCFtyper 进行联合调用的生物信息学流程</center> ``` #Process all samples independently until GVCF for sample in s1 s2 s3; do (sentieon bwa mem -R '@RG\tID:GROUP_${sample}\tSM:${sample}\tPL:PLATFORM' \ -t NUMBER_THREADS REFERENCE ${sample}_FASTQ_1 ${sample}_FASTQ_2 || echo -n \ 'error') | sentieon util sort -o ${sample}_SORTED_BAM -t NUMBER_THREADS \ --sam2bam -i - sentieon driver -t NUMBER_THREADS -i ${sample}_SORTED_BAM \ --algo LocusCollector --fun score_info ${sample}_SCORE_TXT sentieon driver -t NUMBER_THREADS -i SORTED_BAM_1 -i SORTED_BAM_2 \ --algo Dedup --rmdup --score_info ${sample}_SCORE_TXT ${sample}_DEDUPED_BAM sentieon driver -r REFERENCE -t NUMBER_THREADS -i ${sample}_DEDUPED_BAM \ --algo QualCal -k $known_sites ${sample}_RECAL_DATA_TABLE sentieon driver -r REFERENCE -t NUMBER_THREADS -i ${sample}_DEDUPED_BAM \ -q ${sample}_RECAL_DATA_TABLE --algo Haplotyper --emit_mode gvcf \ ${sample}_VARIANT_GVCF done #Run joint variant calling sentieon driver -r REFERENCE --algo GVCFtyper -v s1_VARIANT_GVCF \ -v s2_VARIANT_GVCF -v s3_VARIANT_GVCF VARIANT_VCF ``` - 单独处理每个样本并为每个样本创建重校准的 BAM 文件或去重的 BAM 和重校准表,然后使用 Haplotyper 算法处理所有文件。 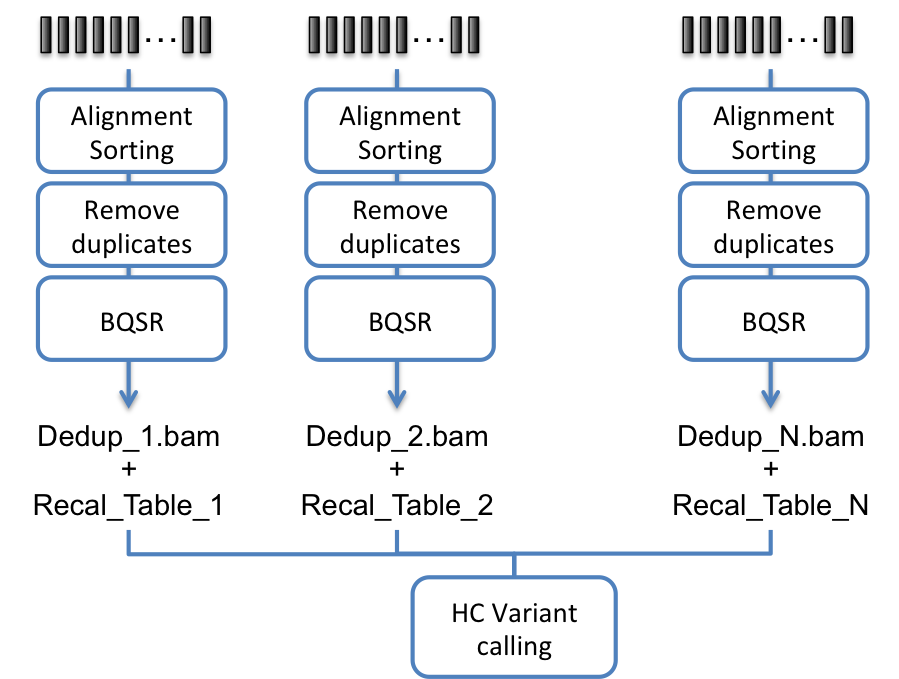 <center>图6-3使用 Haplotyper 和多个 BAM 文件进行联合调用的生物信息学流程</center> ``` #Process all samples independently until BQSR for sample in s1 s2 s3; do (sentieon bwa mem -R '@RG\tID:GROUP_${sample}\tSM:${sample}\tPL:PLATFORM' \ -t NUMBER_THREADS REFERENCE ${sample}_FASTQ_1 ${sample}_FASTQ_2 || echo -n \ 'error' )| sentieon util sort -o ${sample}_SORTED_BAM -t NUMBER_THREADS \ --sam2bam -i - sentieon driver -t NUMBER_THREADS -i ${sample}_SORTED_BAM \ --algo LocusCollector --fun score_info ${sample}_SCORE_TXT sentieon driver -t NUMBER_THREADS -i SORTED_BAM_1 -i SORTED_BAM_2 \ --algo Dedup --rmdup --score_info ${sample}_SCORE_TXT ${sample}_DEDUPED_BAM sentieon driver -r REFERENCE -t NUMBER_THREADS -i ${sample}_DEDUPED_BAM \ --algo QualCal -k $known_sites ${sample}_RECAL_DATA_TABLE done #Run joint variant calling sentieon driver -t NUMBER_THREADS -i s1_DEDUPED_BAM -q s1_RECAL_DATA_TABLE \ -i s2_DEDUPED_BAM -q s2_RECAL_DATA_TABLE -i s3_DEDUPED_BAM \ -q s3_RECAL_DATA_TABLE --algo Haplotyper VARIANT_VCF ``` ## 6.2 Haplotyper 和 Genotyper 中支持的注释 Haplotyper 和 Genotyper 这两种生殖系变异调用方法都接受输出额外注释到结果 VCF 的选项。以下是支持的可能注释: - AC: 基因型中的等位基因计数,针对每个 ALT 等位基因,按列出的顺序 - AF: 等位基因频率,针对每个 ALT 等位基因,按列出的顺序 - AN: 在调用的基因型中的总等位基因数 - BaseQRankSum: Alt 与 Ref 碱基质量的 Wilcoxon 秩和检验的 Z 分数 - ClippingRankSum: Alt 与 Ref 硬裁剪碱基数量的 Wilcoxon 秩和检验的 Z 分数 - DP: 跨样本的组合深度 - ExcessHet: 过度杂合性精确检验的 Phred 标度 p 值 - FS: 使用 Fisher 精确检验检测链偏好的 Phred 标度 p 值 - InbreedingCoeff: 与 Hardy-Weinberg 期望相比,根据每个样本的基因型似然估计的近交系数 - MLEAC: 等位基因计数的最大似然期望 (MLE),针对每个 ALT 等位基因,按列出的顺序 - MLEAF: 等位基因频率的最大似然期望 (MLE),针对每个 ALT 等位基因,按列出的顺序 - MQ: RMS 映射质量 - MQ0: MAPQ == 0 的读数数量 - MQRankSum: Alt 与 Ref 读数映射质量的 Wilcoxon 秩和检验的 Z 分数 - QD: 变异置信度/深度质量 - RAW_MQ: RMS 映射质量的原始数据 - ReadPosRankSum: Alt 与 Ref 读数位置偏好的 Wilcoxon 秩和检验的 Z 分数 - SOR: 检测链偏好的 2x2 列联表的对称比值比 - SAC: 支持每个等位基因(包括参考)的正向和反向链上的读数数量 - AS_BaseQRankSum: 等位基因特异性 - AS_FS: 等位基因特异性使用 Fisher 精确检验检测链偏好的 Phred 标度 p 值 - AS_InbreedingCoeff: 与 Hardy-Weinberg 期望相比,根据每个样本的基因型似然估计的等位基因特异性近交系数 - AS_MQRankSum: 等位基因特异性 Alt 与 Ref 读数映射质量的 Wilcoxon 秩和检验的 Z 分数 - AS_QD: 等位基因特异性变异置信度/深度质量 - AS_MQ: 等位基因特异性 RMS 映射质量 - AS_ReadPosRankSum: 等位基因特异性 Alt 与 Ref 读数位置偏好的 Wilcoxon 秩和检验的 Z 分数 - AS_SOR: 等位基因特异性检测链偏好的 2x2 列联表的对称比值比 ## 6.3 比对后移除低映射质量的读数 以下是比对后如何移除低映射质量的读数的示例,以便它们不会占用输出 BAM 文件的空间。 ``` sentieon bwa mem -R '@RG\tID:GROUP_NAME_1 \tSM:SAMPLE_NAME \tPL:PLATFORM' \ -t NUMBER_THREADS REFERENCE SAMPLE_1 [SAMPLE_2] \ |samtools view -h -q MAP_QUALITY_THRESHOLD - \ | sentieon util sort -o SORTED_BAM_1 -t NUMBER_THREADS --sam2bam -i - ``` ## 6.4 执行 Dedup 以标记主要和非主要读数 标准的双通道 dedup 将忽略非主要读数,并且永远不会改变它们的重复标志。 以下是如何执行 dedup 以同时标记主要和非主要读数的示例: ``` sentieon driver -t NUMBER_THREADS -i SORTED_BAM \ --algo LocusCollector --fun score_info SCORE_TXT sentieon driver -t NUMBER_THREADS -i SORTED_BAM \ --algo Dedup --score_info SCORE_TXT --metrics DEDUP_METRIC_TXT \ -- output_dup_read_name DUPLICATED_READNAMES_TXT sentieon driver -t NUMBER_THREADS -i SORTED_BAM \ --algo Dedup --rmdup --dup_read_name DUPLICATED_READNAMES_TXT DEDUPED_BAM ``` ## 6.5 使用量化质量分数数据时的流程修改 当您的输入数据具有量化的碱基质量分数时,输入 FASTQ 中报告的碱基质量很可能比经验碱基质量高得多;在这种情况下,变异调用步骤可能需要更长时间来处理数据,因为在局部组装中本不会考虑的低质量碱基将导致生成许多不相关的低质量单倍型。这个问题应该通过运行流程的碱基质量分数重校准步骤来解决;或者,您可以修改 Haplotyper 或 TNhaplotyper 的 min_base_qual 参数的默认值以减少影响:在命令行中添加 --min_base_qual 15 选项以防止此问题。 ## 6.6 当肿瘤和正常输入都具有相同的 RGID 时修改 BAM 文件的 RG 信息 如果您输入多个不同的 BAM 文件,其中包含具有相同 ID 但不同属性的读组,Sentieon® 工具将报错。这种情况可能发生在 TNseq® 和 TNscope® 中,当肿瘤和正常样本 BAM 文件的 RG ID 都是 "1" 时。为了解决这个问题,您可以使用该功能来修改 RG 信息。 ``` #first lane tumor, RGID incorrectly set to uninformative 1 sentieon bwa mem -R '@RG\tID:1\tSM:TUMOR_SM\tPL:PLATFORM' -t NT \ REFERENCE TUMOR_1_1.fq.gz TUMOR_1_2.fq.gz | sentieon util sort \ -o TUMOR_1.bam -t NT --sam2bam -i - #second lane tumor, RGID incorrectly set to uninformative 1 sentieon bwa mem -R '@RG\tID:1\tSM:TUMOR_SM\tPL:PLATFORM' -t NT \ REFERENCE TUMOR_2_1.fq.gz TUMOR_2_2.fq.gz | sentieon util sort \ -o TUMOR_2.bam -t NT --sam2bam -i - #normal, RGID incorrectly set to uninformative 1 sentieon bwa mem -R '@RG\tID:1\tSM:NORMAL_SM\tPL:PLATFORM' -t NT \ REFERENCE TUMOR_1.fq.gz TUMOR_2.fq.gz | sentieon util sort \ -o NORMAL.bam -t NT --sam2bam -i - #using replace_rg argument to modify the RGID and make them unique #each replace_RG argument will only affect the following input BAM file sentieon driver -r REFERENCE \ --replace_rg 1='ID:T_1\tSM:TUMOR\tPL:PLATFORM' -i TUMOR_1.bam \ --replace_rg 1='ID:T_2\tSM:TUMOR\tPL:PLATFORM' -i TUMOR_2.bam \ --replace_rg 1='ID:N\tSM:NORMAL\tPL:PLATFORM'-i NORMAL.bam \ --algo TNscope --tumor_sample TUMOR --normal_sample NORMAL OUT_TN_VCF ``` --- # 7 故障排除与常见问题 ## 7.1 故障排除 ### 7.1.1 准备参考文件以供使用 如果您的参考FASTA文件尚未预处理,使得软件无法使用中指定的数据,您需要按照以下步骤对其进行处理: 1. 使用BWA生成BWA索引。这将创建".fasta.amb"、".fasta.ann"、".fasta.bwt"、".fasta.pac"和".fasta.sa"文件。 ``` sentieon bwa index reference.fasta ``` 2. 使用samtools生成FASTA文件索引。这将创建".fasta.fai"文件。 ``` samtools faidx reference.fasta ``` 3. 使用Picard生成序列字典。这将创建".dict"文件。 ``` java -jar picard.jar CreateSequenceDictionary REFERENCE=reference.fasta \ OUTPUT=reference.dict ``` ### 7.1.2 准备RefSeq文件以供使用 RefSeq文件用于将CoverageMetrics算法的结果聚合到基因级别。为了使用从UCSC基因组浏览器下载的RefSeq文件,需要按染色体和位点对它们进行排序。要执行排序,您需要执行以下步骤: 1. 从文件中删除头部 ``` grep -v “^#” FILE.refSeq > FILE.refSeq.headerless grep -e “^#” FILE.refSeq > FILE.refSeq.header ``` 2. 首先使用unix sort对位点进行排序。 ``` sort -k 5 -n FILE.refSeq.headerless > FILE.refSeq.presorted ``` 3. 使用GATK sortByRef.pl(可从获取)和FASTA索引fai按染色体排序。 ``` perl sortByRef.pl --k 3 FILE.refSeq.presorted FASTA.fai --tmp ~/tmp \ > FILE_sorted_headerless.refSeq ``` 4. 将头部放回文件。 ``` cat FILE.refSeq.header > FILE_sorted.refSeq cat FILE_sorted_headerless.refSeq >> FILE_sorted.refSeq ``` ## 7.2 常见问题 ### 7.2.1 Sentieon安装时出现 jemalloc error 在安装运行Sentieon过程中可能会出现以下错误: ``` ERROR: ld.so: object '/usr/lib64/libjemalloc.so.2' from LD_PRELOAD cannot be preloaded: ignored. Failed to contact the license server at 10.10.10.1:8990 ``` 这个错误与jemalloc有关。Jemalloc是一个内存分配器,针对多线程方案中的高内存分配性能和更少的内存碎片进行了优化。Sentieon建议使用jemalloc来改善Sentieon应用程序中的内存管理和整体性能,尤其是Sentieon bwa-mem。 解决方案: 1. 安装jemalloc: 对于不同的操作系统,安装命令如下: - RHEL/CentOS 8.x: ``` yum install epel-release yum install jemalloc ``` 默认安装在 /usr/lib64/libjemalloc.so.2 - RHEL/CentOS 7.x: ``` yum install epel-release yum install jemalloc ``` 默认安装在 /usr/lib64/libjemalloc.so.1 - Ubuntu 20.04: ``` apt update apt install libjemalloc2 ``` 默认安装在 /usr/lib/x86_64-linux-gnu/libjemalloc.so.2 - Ubuntu 18.04: ``` apt update apt install libjemalloc1 ``` 默认安装在 /usr/lib/x86_64-linux-gnu/libjemalloc.so.1 2. 对于没有预构建软件包的其他系统,请参考jemalloc GitHub页面 (https://github.com/jemalloc/jemalloc )以获取有关如何构建和安装jemalloc的更多信息。 3. 使用环境变量在运行时加载jemalloc库到Sentieon中: 例如,在CentOS 8.x系统上,在运行Sentieon工具之前,您可以使用以下命令设置环境变量: ``` export LD_PRELOAD=/usr/lib64/libjemalloc.so.2 ``` ### 7.2.2 许可证消息:No more license available for Sentieon… 当您请求运行Sentieon®软件的线程数超过您的许可证当前允许的数量时,会产生此消息。这种情况发生是因为您同时运行的命令集体请求的线程数超过了您的许可证支持的核心数。可使用以下命令查看授权剩余的线程数: ``` sentieon licclnt query -s LICSRVR_HOST:LICSRVR_PORT klib ``` Sentieon®命令在等待空闲许可证时将处于空闲状态,但命令不会失败。 ### 7.2.3 Driver失败并显示错误:Readgroup XX is present in multiple BAM files with different attributes 当您输入两个不同的BAM文件,其中包含具有相同ID但不同属性的读组时,会产生此错误。例如,在TNseq®和TNscope®中,肿瘤和正常样本BAM文件的RG ID都是"1"。 在使用BAM文件之前,您需要编辑它们以使RG ID唯一,例如通过将SM名称添加到RG ID中。您可以查看此问题的解决方法示例。 或者,您可以使用samtools addreplacerg功能来修改输入BAM文件的RG ID并使其唯一: ``` #add the new RG and modify all reads in the BAM file RGtag=$(samtools view -H $INPUT_BAM|grep ^@RG|sed "s|ID:$ORIG_RGID|ID:$NEW_RGID|g") samtools addreplacerg -r "$RGtag" -o $TMP_BAM $INPUT_BAM #reheader the BAM to remove the original RG that is no longer used samtools view -H $TMP_BAM|grep -v "^@RG.*$ORIGINAL_RGID" \ |samtools reheader - $TMP_BAM > $OUTPUT_BAM rm $TMP_BAM ``` ### 7.2.4 Driver报告警告:none of the QualCal tables is applicable to the input BAM files 此警告意味着重校准表输入文件中的信息都不能应用于输入的BAM文件,这可能是由于使用了与BAM文件不对应的重校准表。 当QualCal的输入BAM文件在RG字段中没有正确的字段时,可能会产生此警告。例如,如果RG的PL标签设置为ILLUMINA以外的其他值,就可能发生这种情况;在这种情况下,您需要修改BAM头以包含/修改缺失/不正确的字段,为此您可以使用samtools reheader功能。 ### 7.2.5 使用从BAM文件创建的FASTQ文件时,BWA使用异常大量的内存 当您使用通过转换已排序的BAM文件创建的FASTQ文件时,可能会发生所有未映射的读段都被分组到FASTQ输入的末尾。在这种情况下,BWA可能在对齐结束时使用异常大量的内存,因为映射质量差或无法映射的读段需要额外的内存。 为了减少异常内存使用,您应该首先重新排序bam文件,以确保未映射的读段不会被分组在一起。您可以使用samtools来做到这一点: ``` samtools sort -n -@ 32 input.bam | samtools fastq -@ 32 \ -s >(gzip -c > single.fastq.gz) -0 >(gzip -c > unpaired.fastq.gz) \ -1 >(gzip -c > output_1.fastq.gz) -2 >(gzip -c > output_2.fastq.gz) - ``` ### 7.2.6 Driver或Util失败并显示错误:can not open file (xxx) in mode(r),Too many open files 此错误的根本原因是系统中同时打开的文件限制设置得不够高。 您可以通过设置系统ulimit -n来解决此错误。在基于Linux的系统中: 检查系统中最大打开文件数的限制,运行以下命令: ``` ulimit -n ``` 通过以root身份编辑文件/etc/security/limits.conf来设置更高的限制,并添加以下2行: ``` * soft nofile 16384 * hard nofile 16384 ``` 如果您的系统运行的是Ubuntu,您还需要将此行添加到您的shell配置文件~/.bashrc中: ``` ulimit -n 16384 ``` 您需要注销系统并重新登录才能使更改生效。登录后,通过运行以下命令检查更改是否正确应用: ``` ulimit -n ``` 命令应返回16384。 ### 7.2.7 Driver失败并显示错误:Contig XXX from vcf/bam is not present in the reference, or error Contig XXX has different size in vcf/bam than in the reference 此错误的根本原因是输入的VCF或BAM文件与参考fasta文件不兼容。文件中的contig不存在于参考中,或contig的大小不同。这很可能是由于使用了与不同参考处理的VCF或BAM文件造成的。 ### 7.2.8 Driver报告警告:Contigs in the vcf file XXX do not match any contigs in the reference 此警告的根本原因是输入的VCF文件与参考fasta文件不兼容,文件中的contig不存在于参考中。这很可能是由于使用了来自不同参考的VCF文件造成的。 ### 7.2.9 BWA失败并显示错误:Killed 当BWA从操作系统接收到SIGKILL信号时,会产生此错误。如果系统可用内存不足,SIGKILL可能是由内核的内存不足(OOM)管理器发送的。您可以检查系统上的内核日志以确认SIGKILL信号是否由OOM管理器发送。 要解决此错误,您可以使用`bwt_max_mem`环境变量减少BWA的内存使用。 ## 7.3 KPNS - 已知问题无解决方案 ### 7.3.1 不支持未使用bgzip压缩的gzip压缩vcf文件 普通的gzip文件不允许随机或索引访问其中包含的信息,只有使用bgzip压缩的文件才可索引。 因此,Sentieon®软件不支持gzip压缩的VCF文件作为输入。 要使用这些文件,您需要使用gunzip解压缩它们,然后不压缩使用或使用bgzip重新压缩。 或者,您可以使用util vcfconvert重新压缩和索引文件。 ``` sentieon util vcfconvert INPUT.vcf.gz OUTPUT.vcf.gz ``` ### 7.3.2 不支持gzip压缩的fasta文件 目前,软件不支持gzip压缩的FASTA文件作为输入。您需要在使用之前gunzip这些文件。 ### 7.3.3 FASTQ文件需要采用SANGER质量格式 如果您的FASTQ文件是在1.8之前使用Illumina™测序技术编码的,读段质量分数将不是SANGER格式,这可能会产生意外结果。Sentieon®基因组学软件不会检测到您正在使用不支持的格式。 ### 7.3.4 Driver失败并显示错误:ImportError: No module named argparse 当在Python版本为2.6.x且不存在argparse模块的环境中运行tnhapfilter时,会产生此错误。您需要为您的Python安装安装argparse模块;您可以通过运行pip install argparse或您使用的其他包管理器来完成此操作。 --- # 8 附录 ## 8.1 缩写和缩略语 GATK:基因组分析工具包(Genome Analysis Tool Kit) VCF:变异检测格式(Variant Call Format) SAM:序列比对/映射格式(Sequence Alignment/Map) BAM:二进制压缩的SAM格式(Binary compressed SAM) BCF:二进制压缩的变异检测格式(Binary compressed Variant Call Format) BED:浏览器可扩展显示格式(Browser Extensible Display) Indels:插入-缺失(INsertion-DELetions) ## 8.2 免责声明和责任限制 SENTIEON公司保留程序交付时的所有权利。未经SENTIEON同意,除许可证规定外,不得以任何形式复制程序或其任何部分。 不得从程序中删除本声明。 SENTIEON、SENTIEON的任何成员、以下组织或代表他们行事的任何人: 1. 不对程序作出任何明示或暗示的保证或陈述,包括对适销性或特定用途适用性的任何保证; 2. 不对程序或其任何部分的任何使用或可能由此产生的任何损害承担任何责任。 在法律允许的最大范围内,除本文明确保证外,SENTIEON提供的软件、文档和其他产品及服务均按"原样"提供,不附带任何种类的明示或暗示保证,包括但不限于所有权保证或对适销性、非侵权或特定用途适用性的暗示保证。SENTIEON及其许可方不保证SENTIEON软件、文档或服务将满足被许可方的要求,无错误或不间断运行。 --- 
chsnp
2026年2月9日 10:31
转发
收藏文档
上一篇
下一篇
手机扫码
复制链接
手机扫一扫转发分享
复制链接
Markdown文件
Word文件
PDF文档
PDF文档(打印)
分享
链接
类型
密码
更新密码
有效期