Sentieon
Sentieon 中文手册
Sentieon 中文手册(上册)
Sentieon 中文手册(下册)
Sentieon 软件应用教程
Sentieon | 应用教程: 使用DNAscope对HiFi长读长数据进行胚系变异检测分析
Sentieon | 应用教程: 利用Sentieon Python API引擎为自研算法加速
Sentieon | 应用教程: 关于读段组的建议
Sentieon | 应用教程: TNscope® 使用机器学习模型进行有匹配正常样本的体细胞变异发现
Sentieon | 应用教程: CCDG使用Sentieon®的功能等效流程
Sentieon | 应用教程: 利用共识功能去除PCR重复
Sentieon | 应用教程: 适用于PacBio HiFi和Oxford Nanopore长读长测序数据的结构变异检测
Sentieon | 应用教程: 使用 Sentieon进行大型基因组重测序分析
Sentieon | 应用教程: 体细胞SNP/Indel变异检测
Sentieon | 应用教程: DNAscope使用机器学习模型进行胚系变异调用
Sentieon | 应用教程: 唯一分子标识符(UMI)
Sentieon | 应用教程: Sentieon分布模式
Sentieon | 应用教程:使用CNVscope进行CNV检测分析
Sentieon发布核心家系(trio)基因分析最佳实践方案
Sentieon推出Segdup-caller:针对片段重复区域的专用精准变异检测工具
Sentieon软件版本更新
Sentieon | 发布V202503.01版本
Sentieon | 发布V202503.02版本
Sentieon软件快速入门指南
Sentieon 软件模块总述
Sentieon 特色流程 - DNAscope
Sentieon | DNAscope Illumina 流程
sentieon | DNAscope Complete Genomics 流程
Sentieon | DNAscope LongRead PacBio 流程
Sentieon | DNAscope Ultima Genomics 流程
Sentieon | DNAscope Element Bio 流程
Sentieon | DNAscope LongRead Nanopore 流程
Sentieon混合分析流程 - DNAscope Hybrid
Sentieon推出混合型短读长和长读长变异检测DNAscope Hybrid流程(上)
Sentieon推出混合型短读长和长读长变异检测DNAscope Hybrid流程(下)
Sentieon | 泛基因组分析流程详解
Sentieon | 物种全基因组(WGS)分析流程
Sentieon | 植物全基因组(GWS)分析流程
Sentieon | 小麦(Triticum_aestivum)全基因组WGS分析流程
Sentieon | 水稻(Oryza_sativa)全基因组WGS分析流程
Sentieon | 拟南芥(Arabidopsis_thaliana)全基因组WGS分析流程
Sentieon | 马铃薯(Solanum_tuberosum)全基因组WGS分析流程
Sentieon | 巨桉(Eucalyptus grandis)全基因组WGS分析流程
sentieon | 向日葵(Helianthus annuus)全基因组WGS分析流程
Sentieon | 野草莓(Fragaria vesca)全基因组WGS分析流程
Sentieon | 银杏(Ginkgo biloba)全基因组WGS分析流程
Sentieon | 大豆(Glycine max)全基因组WGS分析流程
毅硕Sentieon | 陆地棉(Gossypium hirsutum)全基因组WGS分析流程
Sentieon | 动物全基因组(WGS)分析流程
Sentieon | 猪(sus scrofa)全基因组WGS分析流程
Sentieon | 鸡(Gallus gallus)全基因组WGS分析流程
Sentieon | 家鼠(Mus musculus)全基因组WGS分析流程
Sentieon | 家犬(canis lupus familiaris)全基因组WGS分析流程
sentieon | 东方蜜蜂(Apis cerana)全基因组WGS分析流程
Sentieon | 电鳗(Electrophorus electricus)全基因组WGS分析流程
Sentieon | 红隼(Falco tinnunculus)全基因组WGS分析流程
Sentieon | 家猫(Felis catus)全基因组WGS分析流程
Sentieon文献解读
Sentieon文献解读 | Population Sequencing
Sentieon文献解读 | Agrigenomics
Sentieon | Agrigenomics-泛基因组揭示小麦结构变异与栖息地及育种的关联
Sentieon文献解读 | Genetic Disease
Sentieon文献解读 | Tumor Sequencing
Sentieon文献解读 | Benchmark and Method Study
Sentieon文献解读 | Long Read Sequencing
Sentieon文献解读 | Clinical Trial
Sentieon文献解读 | Epidemiology
Sentieon文献解读 | Gene Editing
Sentieon文献解读 | Liquid Biopsy
-
+
首页
Sentieon 中文手册(上册)
# 1 Sentieon简介    Sentieon软件为完整的纯软件基因变异检测二级分析方案,其分析流程完全忠于BWA/GATK/MuTect2/STAR/ Minimap2/ Fgbio/picard等金标准的数学模型。在匹配开源流程分析结果的前提下,大幅提升WGS、WES、Panel、UMI、ctDNA、RNA等测序数据的分析效率和检出精度,并匹配目前全部第二代、三代测序平台,对短读长NGS、长读长测序数据进行SNP/INDEL/SV分析。  <center>图1-1 Sentieon 基因组学软件</center>    Sentieon为纯CPU计算加速软件,完全适配主流CPU计算架构:Intel、AMD、海光等X86架构CPU,华为鲲鹏、阿里倚天、Ampare、Apple Silicon等ARM架构CPU。可灵活部署在单机工作站、HPC集群、超算中心和云计算中心,保持同一套流程下不同规模数据计算结果的一致性。    Sentieon软件团队拥有丰富的软件开发及算法优化工程经验,致力于解决生物数据分析中的速度与准确度瓶颈,为来自于分子诊断、药物研发、临床医疗、人群队列、动植物等多个领域的合作伙伴提供高效精准的软件解决方案,共同推动基因技术的发展。    截至2025年7月份,Sentieon已经在全球范围内为1860+用户提供服务,用户处理超过4980+PB数据量,被世界一级影响因子刊物如NEJM、Cell、Nature等广泛引用,引用次数超过1500篇。此外,Sentieon连续数年摘得了Precision FDA、Dream Challenges等多个权威评比的桂冠,在业内获得广泛认可。 --- # 2 Sentieon快速入门指南 本指南旨在帮助初次使用 Sentieon® 软件的用户快速上手。如果您有任何其他疑问,请通过 support@sentieon.com 联系 Sentieon® 公司的技术支持团队。 ## 2.1 运行环境 ### 2.1.1 硬件要求 Sentieon Genomics 软件专为 Linux 及其他符合 POSIX 标准的平台设计,确保了在高性能计算环境下的卓越兼容性。 对于 Linux 系统,我们建议使用具有以下配置的Linux服务器: * 运行以下任一发行版或更高版本的 Linux 系统:RedHat/CentOS 6.5、Debian 7.7、OpenSUSE-13.2或Ubuntu-14.04。 * 处理小型Panel或全外显子组至少需要16GB内存,处理全基因组至少需要64GB内存。 * (推荐)首选高速SSD硬盘,以获得理想的I/O性能并实现CPU利用率的最大化。 ### 2.1.2 软件要求 系统需要安装Python 2.6.x、Python 2.7.x或python3.x。您可以通过输入以下命令检查Python版本: ``` python --version ``` ### 2.1.3 Sentieon®软件发布包 从Sentieon技术支持提供的链接下载软件包。 - https://ftp.insvast.com/user/Sentieon/release/sentieon-genomics-202503.02.tar.gz (适配X86架构CPU服务器,例如Intel、 AMD、 曙光) - https://ftp.insvast.com/user/Sentieon/release/arm-sentieon-genomics-202503.02.tar.gz (适配ARM架构CPU服务器, 例如华为鲲鹏、 阿⾥倚天、 Ampere) - 软件包下载链接用户名:insvast;密码:Ins@1234; 通过运行以下命令解压软件包,其中VERSION是您使用的版本,例如202503.02: ``` tar xvzf sentieon-genomics-VERSION.tar.gz ``` ### 2.1.4 授权许可(License)要求 Sentieon®软件是一个商用许可的软件。用户需要正确设置授权许可才能运行软件。我们提供两种类型的授权许可: * **单机评估授权许可**:此类授权许可用于在单台机器上评估试用Sentieon®软件。它允许新用户在无需 IT 部门协助的情况下快速上手使用软件。要使用此类授权许可,计划运行Sentieon®软件的计算机需要具备外部Internet访问权限。 * **集群授权许可**:此类授权许可用于集群环境。使用此类授权许可,一个轻量级的浮动授权许可服务器进程在集群中的某个节点上运行,并通过TCP协议向所有其他与授权许可服务器有网络连接的节点提供授权服务。此类授权许可服务器通常在集群外围的一个特殊的非计算节点上运行,该节点可以通过HTTPS不受限制地访问外网,并通过监听集群内需要开放的特定TCP端口向集群中的其余节点提供授权。 #### (1)设置单机评估授权许可 要使用单机评估授权许可,计算节点需要能够访问Internet。以便Sentieon®软件验证授权许可。 要使用单机评估授权许可,请按照以下步骤操作: 1. 将授权许可文件复制到计算节点。例如,许可证文件LICENSE_FILE.lic现在位于LICENSE_DIR目录下。 1. 按如下方式设置环境变量: ``` export SENTIEON_LICENSE=LICENSE_DIR/LICENSE_FILE.lic ``` #### (2)设置授权许可服务器 如图2-1所示,授权许可服务器需要满足以下条件: 1. 授权许可服务器应该能够访问Internet,以便执行授权许可验证。 3. 计算节点应该能够通过主机名`LICSRVR_HOST`访问授权许可服务器。 4. 运行许可证服务器的机器需要开放一个端口供授权许可服务进行监听,且计算节点可以访问该端口。这里我们假设可用端口是`LICSRVR_PORT`。 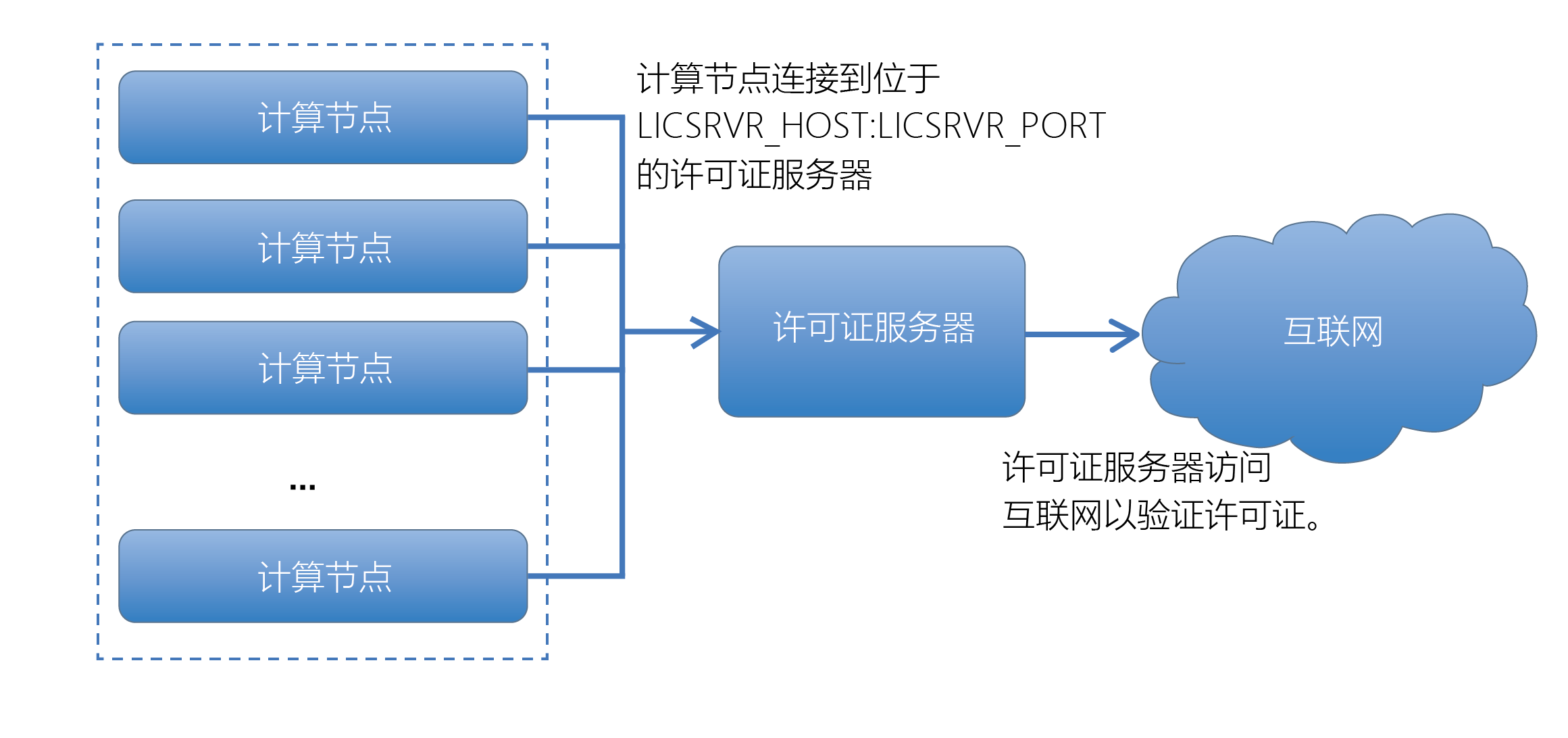 <center>图2-1 计算节点和授权许可服务器的拓扑结构</center> <br> 您可能需要IT部门的支持来获取`LICSRVR_HOST:LICSRVR_PORT`参数,并确认已满足上述要求。 **注意:** 如果授权许可服务器位于防火墙后面,并通过NAT与计算节点分离,那么计算节点可见的授权许可服务器主机名/IP可能与实际主机名/IP不同。如果是这种情况,您需要将授权许可服务器绑定到实际IP地址上,而计算节点则需通过 NAT 后的 IP 地址请求授权许可。详情请联系 Sentieon 技术支持。 按照以下步骤获取授权许可文件,设置和测试授权许可服务器: 1. 将以下信息发送给sentieon@insvast.com以获取授权许可文件: * 指定运行授权许可服务的机器的FQDN(Hostname),即`LICSRVR_HOST`。 * 指定的监听端口`LICSRVR_PORT`。 2. 将收到的授权许可文件复制到授权许可服务器`LICSRVR_HOST`上。我们假设授权许可文件位于`LICENSE_PATH/LICENSE_FILE`。在授权许可服务器上运行以下命令来启动授权许可服务器进程: ``` <SENTIEON_INSTALL_DIR>/bin/sentieon licsrvr --start --log LOG_FILE LICENSE_PATH/LICENSE_FILE ``` 3. 或者,您可以按照 *“2.3设置许可为系统服务”* - 将授权许可服务器(LICSRVR)作为系统服务运行的说明,将授权许可服务器配置并作为系统守护进程启动。 5. 进入Sentieon®安装目录。在授权许可服务器上运行以下命令,以确认授权许可服务器已正常启动并运行。 ``` <SENTIEON_INSTALL_DIR>/bin/sentieon licclnt ping -s LICSRVR_HOST:LICSRVR_PORT ``` 如果命令返回时没有显示错误信息,则表明授权许可服务器已启动并成功运行。 5. 登录到其中一个计算节点,进入Sentieon®安装目录,再次运行上述命令: ``` <SENTIEON_INSTALL_DIR>/bin/sentieon licclnt ping -s LICSRVR_HOST:LICSRVR_PORT ``` 如果该命令返回时没有显示错误信息,则表明计算节点现在也可以访问授权许可服务器了。 6. 设置以下环境变量,您就可以开始使用Sentieon了。 ``` export SENTIEON_LICENSE=LICSRVR_HOST:LICSRVR_PORT ``` ### 2.1.5 系统环境要求 - 如果Python 2.6.x、Python 2.7.x或python3.x不是默认Python版本,您可以设置以下环境变量: ``` export SENTIEON_PYTHON=Python_location ``` - 如果您使用本地许可证文件,请设置以下环境变量,其中LICENSE_DIR是许可证文件所在的目录,LICENSE_FILE.lic是许可证文件名: ``` export SENTIEON_LICENSE=LICENSE_DIR/LICENSE_FILE.lic ``` - 如果您使用的是许可证服务器,请设置以下环境变量,其中LICSRVR_HOST和LICSRVR_PORT分别是许可证服务器的主机名和端口。详情请参阅下一节。 ``` export SENTIEON_LICENSE=LICSRVR_HOST:LICSRVR_PORT ``` - 为方便调用,请按如下所示设置二进制程序路径,其PATH_TO_SENTIEON_BINARY_DIRECTORY是Sentieon®二进制文件的安装目录: ``` Export SENTIEON_INSTALL_DIR=PATH_TO_SENTIEON_BINARY_DIRECTORY ``` - 在使用NFS(网络文件系统)存储时,为提升性能,请将SENTIEON_TMPDIR环境变量设置为指向本地的高速暂存盘: ``` export SENTIEON_TMPDIR=/tmp ``` ## 2.2 首次运行作业 我们提供了一个快速入门包,其中包含示例脚本和数据,以帮助您快速测试安装并诊断潜在问题。 quikstart演示项目链接: [https://ftp.insvast.com/sentieon\_quickstart.tar.gz](https://ftp.insvast.com/sentieon_quickstart.tar.gz) 快速入门包包含单个染色体的数据,包括样本的序列数据和参考材料。该任务脚本使用Sentieon DNAscope流程处理一组双端Illumina fastq文件,具体步骤包括: * BWA:将读段比对到参考基因组。 * Metrics和LocusCollector:收集读段的统计信息。 * Dedup:重复序列标记(去重)。 * Variant calling:使用DNAscope进行变异检测。 **注意:** DNAscope仅推荐用于二倍体生物的样本。对于其他类型的样本,请使用DNAseq流程。 ### 2.2.1运行快速入门包 开始使用前,请将下载的快速入门包复制到一个新目录,并通过运行以下命令进行解压: ``` tar xzvf sentieon_quickstart.tar.gz ``` 该软件包中包含以下内容: * sentieon_quickstart.sh: 驱动整个流程的示例shell脚本。 * reference: 包含人类基因组参考文件和已知SNP位点数据库文件的目录。 * models: 包含DNAscope模型文件的目录。 * FASTQ files: 样本序列文件。 在运行脚本之前,您需要确保已正确设置了上述环境变量,包括授权许可和软件路径。 然后使用您喜欢的编辑器修改sentieon_quickstart.sh中的用户设置部分: ``` # Update with the location of the Sentieon software package SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-202503.02 # Update with the location of temporary fast storage and uncomment #SENTIEON_TMPDIR=/tmp # It is important to assign meaningful names in actual cases. # It is particularly important to assign different read group names. sample="sample_name" group="read_group_name" platform="ILLUMINA" # Other settings nt=16 #number of threads to use in computation # Is the data prepared with a PCR free library prep PCRFREE=true ``` **注意:** 在用户设置脚本 `sentieon_quickstart.sh` 中: * 在实际情况下,指定具有明确意义的名称非常重要。 * 为不同的数据指定不同的读组名称尤为重要。 用户可以运行以下 `nproc` 命令来获取系统可用的 CPU 核心数: ``` nproc ``` 为了更好地理解`sentieon_quickstart.sh`脚本的其余部分,请阅读每个部分的注释以及手册中的相应章节。 现在,只需运行`sentieon_quickstart.sh`来启动脚本,并观察结果生成。在典型的Linux服务器上,整个运行过程大约需要3-5分钟。实际耗时则取决于您的计算环境。 ``` sh sentieon_quickstart.sh & ``` ### 2.2.2 首次运行作业的输出文件 下是输出文件列表、其代表的含义及参考说明。更多详情请参阅完整文档。 |文件名|描述| |--|--| |sorted.bam|使用 Sentieon® BWA mem 进行比对后,并按基因组坐标排序的 BAM 文件| |score.txt|重复序列的数据文件| |aln_metrics.txt|针对双端测序数据的比对情况及常规统计信息| |gc_summary.txt|GC 偏好性统计摘要| |gc_metrics.txt qc-report.pdf|GC 偏好性统计原始数据及 PDF 报告| |qd_metrics.txt qd-report.pdf|碱基质量分值分布数据文件及PDF报告| |mq_metrics.txt mq-report.pdf|平均质量分值随测序循环变化的数据文件及PDF报告| |is_metrics.txt is-report.pdf|插入片段大小分布的数据文件及PDF报告| |deduped.bam|去重阶段生成的BAM 文件,已移除重复序列| |dnascope.vcf.gz|DNAscope 变异检测生成的压缩格式 VCF 文件| ## 2.3 设置许可为系统服务 ### 2.3.1 使用 sysvinit 将许可证服务器作为系统服务运行 如果您的系统遵循传统的 System V启动脚本,您可以通过以 root 身份运行以下命令来设置许可证服务器在系统中自动启动: 1. 创建并自定义配置文件;配置文件通常为 /etc/sysconfig/licsrvr;但在 Ubuntu 中,配置文件将是 /etc/default/licsrvr。以下是配置文件的示例,使用推荐的用户名 sentieon 设置: - /home/sentieon/release/latest 是指向最新 Sentieon® 软件包安装目录的符号链接 - /home/sentieon/licsrvr 是运行 licsrvr 服务的文件夹 - /home/sentieon/licsrvr/licsrvr.lic 是 Sentieon® 许可证文件 ``` licsrvr="/home/sentieon/release/latest/bin/sentieon licsrvr" licfile=/home/sentieon/licsrvr/licsrvr.lic logfile=/home/sentieon/licsrvr/licsrvr.log ``` 2. 将许可证服务器启动脚本安装到 /etc/init.d 目录中。启动脚本包含在发布包中。 ``` install -m 0755 $SENTIEON_INSTALL_DIR/doc/licsrvr.sh /etc/init.d/licsrvr ``` 3. 安装并启用服务。根据您的系统,您将运行不同的命令: - 如果您的系统安装了 Linux 标准基础核心规范,执行系统 init 脚本安装脚本。 ``` /usr/lib/lsb/install_initd /etc/init.d/licsrvr ``` - 如果您的系统未安装 lsb 一致性包,使用 chkconfig 命令启用服务。 ``` chkconfig --add licsrvr chkconfig licsrvr on ``` - 对于 Ubuntu 和 Debian 系统,如果您没有 lsb/install_initd 二进制文件并选择不安装 lsb-core 包,使用 update-rc.d 命令安装并启用服务。 ``` update-rc.d licsrvr defaults update-rc.d licsrvr enable ``` 4. 您可以使用 service 命令来启动/停止/重启/检查服务状态。 ``` service licsrvr [start|stop|restart|status] ``` ### 2.3.2 使用 systemd 将许可证服务器作为系统服务运行 您可以使用操作系统的 systemd 系统和服务功能来设置许可证服务器在系统中自动启动。为此,以 root 身份运行以下命令: 1. 如果您使用 Sentieon® 软件发布包的 doc 文件夹中的 licsrvr.service 许可证服务器启动脚本,您需要创建脚本所需的必要文件,包括使用用户名 sentieon: * /home/sentieon/release/latest 是指向最新 Sentieon® 软件包安装目录的符号链接 * /home/sentieon/licsrvr 是运行 licsrvr 服务的文件夹 * /home/sentieon/licsrvr/licsrvr.lic 是 Sentieon® 许可证文件 或者,您可以编辑许可证服务器启动脚本以指向您特定的用户名和/或文件位置信息。 2. 将许可证服务器启动脚本安装到 /etc/systemd/system 目录中。 ``` install -m 0644 $SENTIEON_INSTALL_DIR/doc/licsrvr.service /etc/systemd/system ``` 3. 运行以下命令以启用计算机启动时自动启动许可证服务器: ``` systemctl enable licsrvr.service ``` 4. 您可以使用 systemctl 命令手动启动和停止服务。 ``` systemctl start licsrvr.service systemctl stop licsrvr.service ``` --- # 3 Sentieon工具集 下表展示了 Sentieon® 的不同产品系列、工具及其用途。同时,表中还注明了各工具是否实现了与现有 GATK 流程工具等效的功能。 ## 3.1 Sentieon工具列表 |Sentieon®产品| Sentieon®工具| 典型用途| 等效的GATK流程工具| | --- | --- | --- |--- | |Sentieon® BWA |Sentieon® BWA| 读段比对和映射 |BWA| |DNAscope| DNAscope |改进的胚系SNV/Indel/SV检测| -| |DNAseq® | Genotyper| 胚系SNV/Indel检测,基于非单倍型的算法 |UnifiedGenotyper| |DNAseq®| Haplotyper | 胚系SNV/Indel检测 | HaplotypeCaller | |DNAseq® | GVCFtyper | 群体联合分型,已验证可支持多达200,000个样本| GenotypeGVCFs| |DNAseq® |VarCal | 计算变异质量分数重校准 | VariantRecalibrator | |DNAseq® | ApplyVarCal| 应用变异质量分数重校准 | ApplyRecalibration| |RNAseq| RNASplitReadsAtJunction | RNA SNV/Indel检测 |SplitNCigarReads | | RNAseq | Haplotyper | RNA SNV/Indel检测| HaplotypeCaller| |TNseq®| TNsnv |体细胞SNV检测,基于非单倍型的算法 |MuTect| |TNseq®| TNhaplotyper |体细胞SNV/Indel检测| MuTect2| |TNseq® |TNhaplotyper2 + TNfilter |体细胞SNV/Indel检测| GATK4中的Mutect2和FilterMutectCalls| |TNscope® |TNscope® |改进的体细胞SNV/Indel/SV检测| -| |通用工具| Dedup和LocusCollector |执行重复序列标记/去重| Picard MarkDuplicates| |通用工具| Realigner |为基于非单倍型算法的变异检测工具执行Indel重比对| RealignerTargetCreator和IndelRealigner| |通用工具 |QualCal |执行碱基质量分数重校准 |BaseRecalibrator、 AnalyzeCovariates| |通用工具| ReadWriter| 生成BAM文件| PrintReads| |通用工具 |AlignmentStat| QC指标 |Picard CollectAlignmentSummaryMetrics| |通用工具 |BaseDistributionByCycle| QC指标 |Picard CollectBaseDistributionByCycle| |通用工具 |CollectVCMetrics |QC指标| Picard CollectVariantCallingMetrics| |通用工具| ContaminationAssessment| QC指标| ContEst| |通用工具| CoverageMetrics |QC指标| DepthOfCoverage| |通用工具| GCBias| QC指标| Picard CollectGcBiasMetrics| |通用工具| HsMetricAlgo |QC指标 |Picard CollectHsMetrics| |通用工具| InsertSizeMetricAlgo |QC指标| Picard CollectInsertSizeMetrics| |通用工具 |MeanQualityByCycle| QC指标| Picard MeanQualityByCycle| |通用工具 |QualDistribution |QC指标 |Picard QualityScoreDistribution| |通用工具 |QualityYield |QC指标| Picard CollectQualityYieldMetrics |通用工具| SequenceArtifactMetricsAlgo |QC指标| PicardCollectSequencingArtifactMetric, ConvertSequencingArtifactToOxoG| |通用工具| WgsMetricsAlgo |QC指标 |PicardCollectWgsMetrics| --- # 4 Sentieon典型工作流程 ## 4.1 DNAseq® Sentieon® Genomics软件的一个典型用途,是执行Broad研究所最佳实践中推荐的DNA分析生物信息学流程,详见https://www.broadinstitute.org/gatk/guide/best-practices 。图4-1展示了这样一个典型的生物信息学流程。 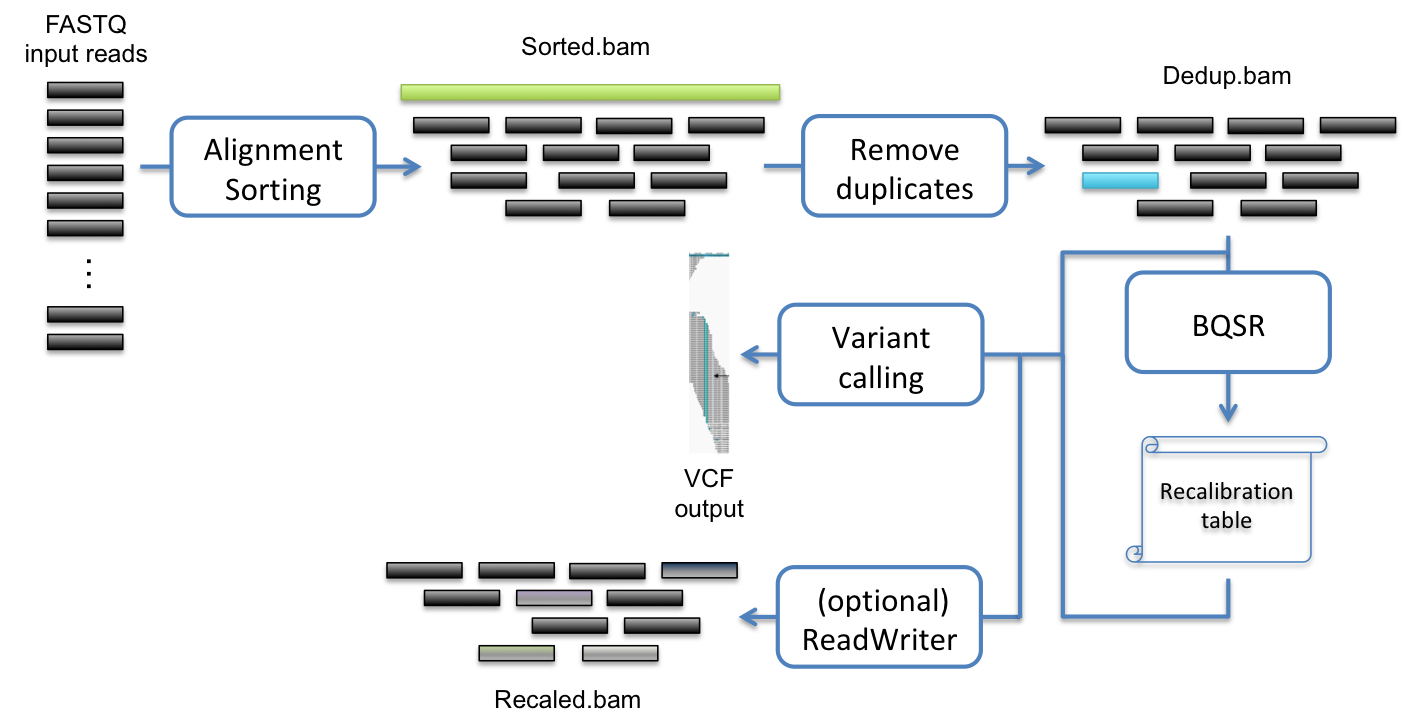 <center>图4-1 推荐的 DNA 变异检测分析的生物信息学流程</center> ### 4.1.1 概述 在这个生信分析流程中,您需要准备以下输入文件: - FASTA文件:包含与您将分析的样本相对应的参考基因组核苷酸序列。参考数据需要进行预处理,以便软件可以使用*表4-1参考核苷酸序列的数据要求*中指定的数据。您可以参考"准备参考文件以供使用"的说明来了解如何生成所需的文件。 <center>表 4-1 参考核苷酸序列的数据要求</center> 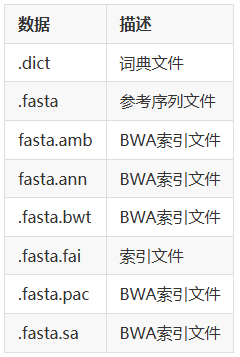 - FASTQ文件:一个或多个包含待分析样本核苷酸序列的文件。这些文件包含来自DNA测序的原始读数。软件支持输入使用GZIP压缩的FASTQ文件。软件仅支持包含Sanger格式(Phred+33)质量分数的文件。 - (可选)您希望包含在流程中的单核苷酸多态性数据库(dbSNP)数据。数据以 VCF 文件的形式使用;您可以使用经过 bgzip 压缩并建立索引的 VCF 文件。 - (可选)您希望包含在流程中的多个已知位点集合。数据以VCF文件的形式使用;您可以使用经过bgzip压缩并索引的VCF文件。 典型的生信分析流程包括以下步骤: 1. 将读数映射到参考基因组:此步骤将 FASTQ 文件中的读段比对并映射到 FASTA 文件中的参考基因组上。该步骤确保了数据能够被置于基因组上下文背景中(确定其位置)。 2. 计算数据指标:此步骤生成关于数据质量和流程分析质量的统计摘要。 3. 去除或标记重复序列:此步骤检测表明同一DNA分子被多次测序的读数。这些重复序列不具有信息性,不应作为额外的证据进行计数。 4. (可选)碱基质量分数重校准(BQSR):此步骤修正测序读段数据中单个碱基的质量分数。此操作可消除由测序方法学产生的实验偏好性。 6. 变异检测:此步骤识别数据中相对于参考基因组出现变异的位点,并计算每个样本在这些位点上的基因型。 ### 4.1.2 DNAseq®使用步骤 #### (1)将读数映射到参考基因组 运行单个命令以高效地使用BWA执行比对,以及使用Sentieon®软件创建BAM文件并进行排序: ``` (sentieon bwa mem -R '@RG\tID:GROUP_NAME\tSM:SAMPLE_NAME\tPL:PLATFORM' \ -t NUMBER_THREADS REFERENCE FASTQ [FASTQ2] || echo -n 'error' ) \ | sentieon util sort -r REFERENCE -o SORTED_BAM -t NUMBER_THREADS --sam2bam -i - ``` BWA的输入和选项在手册中有详细说明。 此外,您也可以使用其他能在`stdout`中生成遵循SAM格式的文件的比对器,并替换命令中的BWA部分。 运行该命令需要以下输入参数: - `GROUP_NAME`:将添加到读组头行的读组标识符。RG:ID在所有您计划使用的所有数据集中必须是唯一的,这在处理多个输入文件或执行肿瘤-正常对照分析时尤为重要。 - `SAMPLE_NAME`:将添加到读组头行的样本名称。 - `PLATFORM`:用于测序DNA的测序平台名称。可能的选项有:ILLUMINA(当fastq文件在Illumina™机器上产生时);IONTORRENT(当fastq文件在Life Technologies™ Ion-Torrent™机器上产生时);ELEMENT(当fastq文件在Element Biosciences™机器上产生时);DNBSEQ(当fastq文件在MGI™机器上产生时);ULTIMA(当fastq文件在Ultima Genomics™机器上产生时)。 - `NUMBER_THREADS`:计算中使用的线程数。我们建议该数量不要超过系统中可用的计算核心数。我们建议BWA和util二进制文件使用相同数量的线程。 - `REFERENCE`:参考FASTA文件的路径。您应确保*表4-1*中指定的所有额外参考数据都在同一位置,并具有一致的命名。 - `FASTQ`:样本FASTQ文件的路径。如果数据来自双端测序技术,您还需要输入FASTQ2作为相应的配对样本FASTQ文件。 - `SORTED_BAM`:映射并排序后的 BAM 输出文件的路径和文件名。系统将同时创建一个对应的索引文件(.bai)。 BWA会根据命令中使用的线程数产生略微不同的比对结果。这是因为 BWA 是基于数据块来计算插入片段大小分布的,而数据块的大小取决于线程数。为了保证结果与使用的线程数无关,您应该使用选项`-K 10000000`来固定数据块大小(以碱基为单位)。 #### (2)计算数据指标 运行单条命令以生成5项关于数据质量和流程分析质量结果的统计摘要: ``` sentieon driver -t NUMBER_THREADS -r REFERENCE -i SORTED_BAM \ --algo GCBias --summary GC_SUMMARY_TXT GC_METRIC_TXT \ --algo MeanQualityByCycle MQ_METRIC_TXT \ --algo QualDistribution QD_METRIC_TXT \ --algo InsertSizeMetricAlgo IS_METRIC_TXT \ --algo AlignmentStat ALN_METRIC_TXT ``` 运行以下四条命令以从统计摘要中生成图表: ``` sentieon plot GCBias -o GC_METRIC_PDF GC_METRIC_TXT sentieon plot MeanQualityByCycle -o MQ_METRIC_PDF MQ_METRIC_TXT sentieon plot QualDistribution -o QD_METRIC_PDF QD_METRIC_TXT sentieon plot InsertSizeMetricAlgo -o IS_METRIC_PDF IS_METRIC_TXT ``` 这些命令需要以下输入参数: - `NUMBER_THREADS`:计算中将使用的线程数。建议该数量不要超过系统中可用的计算核心数。 - `REFERENCE`:参考FASTA文件的路径。需确保参考序列与映射阶段使用的完全一致。 - `SORTED_BAM`:前一个映射阶段存储结果的路径。 - `GC_SUMMARY_TXT`:GC偏差指标摘要结果输出文件的路径和文件名。 - `GC_METRIC_TXT`:GC偏差指标结果输出文件的路径和文件名。 - `MQ_METRIC_TXT`:映射质量指标结果输出文件的路径和文件名。 - `QD_METRIC_TXT`:质量/深度指标结果输出文件的路径和文件名。 - `IS_METRIC_TXT`:插入片段大小指标结果输出文件的路径和文件名。 - `ALN_METRIC_TXT`:比对指标结果输出文件的路径和文件名。 - `GC_METRIC_PDF`:GC偏差指标报告输出文件的路径和文件名。 - `MQ_METRIC_PDF`:映射质量指标报告输出文件的路径和文件名。 - `QD_METRIC_PDF`:质量/深度指标报告输出文件的路径和文件名。 - `IS_METRIC_PDF`:插入片段大小指标报告输出文件的路径和文件名。 #### (3)去除或标记重复序列 在比对和排序完成后,通过两条独立命令对 BAM 文件执行重复序列的移除或标记。第一条命令收集 读段信息,第二条命令执行去重;`--rmdup` 选项控制是直接移除重复读段还是仅将其标记为重复。 ``` sentieon driver -t NUMBER_THREADS -i SORTED_BAM \ --algo LocusCollector --fun score_info SCORE.gz sentieon driver -t NUMBER_THREADS -i SORTED_BAM \ --algo Dedup [--rmdup] --score_info SCORE.gz \ --metrics DEDUP_METRIC_TXT DEDUPED_BAM ``` 这些命令需要以下输入参数: - `NUMBER_THREADS`:计算中将使用的线程数。我们建议该数量不要超过系统中可用的计算核心数。 - `SORTED_BAM`:前一个映射阶段存储结果的路径。 - `SCORE.gz`:临时分值输出文件的路径和文件名。务必确保两条命令使用的是同一个文件。 - `DEDUP_METRICS_TXT`:去重指标结果输出文件的路径和文件名。 - `DEDUPED_BAM`:去重BAM输出文件的路径和文件名。系统将创建相应的索引文件(.bai)。 #### (4)碱基质量分数重校准(BQSR;可选) 运行单条命令来计算序列测序数据中单个碱基质量分数所需的修正值;实际的重校准应用将在变异检测阶段执行。 ``` sentieon driver -t NUMBER_THREADS -r REFERENCE \ -i DEDUPED_BAM --algo QualCal [-k KNOWN_SITES] RECAL_DATA.TABLE ``` 运行三条命令来应用重校准并生成碱基质量分数重校准的报告。第一条命令应用重校准以计算校准后的数据表,并额外对BAM文件执行重校准;第二条命令生成用于绘图的数据;第三条命令将校准前后的数据表绘制成PDF中的图表。 ``` sentieon driver -t NUMBER_THREADS -r REFERENCE -i DEDUPED_BAM \ -q RECAL_DATA.TABLE --algo QualCal [-k KNOWN_SITES] \ RECAL_DATA.TABLE.POST [--algo ReadWriter RECALIBRATED_BAM] sentieon driver -t NUMBER_THREADS --algo QualCal --plot \ --before RECAL_DATA.TABLE --after RECAL_DATA.TABLE.POST RECAL_RESULT.CSV sentieon plot QualCal -o BQSR_PDF RECAL_RESULT.CSV ``` 这些命令需要以下输入参数: - `NUMBER_THREADS`:计算中将使用的线程数。我们建议该数量不要超过系统中可用的计算核心数。 - `REFERENCE`:参考FASTA文件的路径及文件名。需确保参考序列与映射阶段使用的完全一致。 - `DEDUPED_BAM`:前一去重阶段存储结果的路径和文件名。 - `RECAL_DATA.TABLE`:重校准表的路径和文件名。 - `RECAL_DATA.TABLE.POST`:生成的临时校准后重校准表的路径和文件名。 - `RECAL_RESULT.CSV`:用于绘图的临时重校准结果输出文件的路径和文件名。 - `BQSR_PDF`:BSQR结果输出文件的路径和文件名。 以下输入是可选的参数: - `KNOWN_SITES`:用作已知位点集合的VCF文件的路径及文件。您可以通过重复使用`-k KNOWN_SITES`选项来包含多个已知位点集合。 - `RECALIBRATED_BAM`:重校准BAM输出文件的路径和文件名。系统将创建相应的索引文件(.bai)。这个输出是可选的,因为Sentieon®变异检测工具可以使用重校准前的BAM文件配合重校准表,实时执行重校准。 #### (5)变异检测 运行单条命令来检测变异,并同时应用此前计算得到的BQSR。 ``` sentieon driver -t NUMBER_THREADS -r REFERENCE -i DEDUPED_BAM \ -q RECAL_DATA.TABLE --algo Haplotyper [-d dbSNP] VARIANT_VCF ``` 您可能只想重新运行变异检测,例如使用Genotyper(基于位点算法,等效于 Unified Genotyper)变异检测算法。在这种情况下,您不需要重新应用BQSR,可以直接使用之前生成的已重校准的BAM文件: ``` sentieon driver -t NUMBER_THREADS -r REFERENCE -i RECALIBRATED_BAM \ --algo Genotyper [-d dbSNP] VARIANT_VCF ``` 在这两种情况下,使用重校准的BAM或重校准前的BAM加上重校准数据表,都会得到相同的结果;但是,您应当注意不要将重校准数据表与已经重校准过的BAM一起使用,因为这会导致重校准被应用两次,从而导致不正确的结果。 这些命令需要以下输入参数: * `NUMBER_THREADS`:计算中将使用的线程数。我们建议该数量不要超过系统中可用的计算核心数。 * `REFERENCE`:参考FASTA文件的路径及文件名。需确保参考序列与映射阶段使用的完全一致。 * `DEDUPED_BAM`:前一去重阶段存储结果的路径和文件名。 * `RECAL_DATA.TABLE`:前一个BQSR阶段存储结果的路径和文件名。 * `RECALIBRATED_BAM`:已重校准BAM文件的路径。 * `VARIANT_VCF`:变异检测输出文件的路径和文件名。系统将创建相应的索引文件。如果使用 `.gz` 扩展名,工具将输出压缩文件。 以下输入是可选的: * `dbSNP`:将用于标记已知变异的单核苷酸多态性数据库(dbSNP)的路径和文件名。您只能使用一个dbSNP文件。 ### 4.1.3 DNAseq® CCDG标准分析脚本 以下是遵循CCDG标准化流程的DNA测序的Sentieon DNAseq分析脚本示例: ``` #!/bin/sh # Copyright (c) 2016-2020 Sentieon Inc. All rights reserved # ********************************************************************************* # Script to perform DNA seq variant calling using Sentieon following # the functional equivalent pipeline described in # https://github.com/CCDG/Pipeline-Standardization/blob/master/PipelineStandard.md # ********************************************************************************* set -eu # Update with the fullpath location of your sample FASTQ SM="sample" #sample name RGID="rg_$SM" #read group ID PL="ILLUMINA" #or other sequencing platform FASTQ_1="${SM}_r1.fastq.gz" FASTQ_2="${SM}_r2.fastq.gz" #if using 2 FASTQ inputs # Update with the location of the reference data files FASTA_DIR="/home/regression/references/hg38bundle" FASTA="$FASTA_DIR/Homo_sapiens_assembly38.fasta" KNOWN_DBSNP="$FASTA_DIR/Homo_sapiens_assembly38.dbsnp138.vcf.gz" KNOWN_INDELS="$FASTA_DIR/Homo_sapiens_assembly38.known_indels.vcf.gz" KNOWN_MILLS="$FASTA_DIR/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz" # Update with the location of the Sentieon software package and license file SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-|release_version| export SENTIEON_LICENSE=/home/Licenses/Sentieon.lic #or using licsrvr: c1n11.sentieon.com:5443 # Other settings NT=$(nproc) #number of threads to use in computation SAMBLASTER=/home/release/other_tools/samblaster-0.1.23/samblaster START_DIR="$PWD/test/CCDG" #Determine where the output files will be stored # You do not need to modify any of the lines below unless you want to tweak the pipeline # ************************************************************************************************************************************************************************ # ****************************************** # 0. Setup # ****************************************** WORKDIR="$START_DIR/${SM}" mkdir -p $WORKDIR LOGFILE=$WORKDIR/run.log exec >$LOGFILE 2>&1 cd $WORKDIR # ****************************************** # 1. Mapping BWA-MEM 0.7.15 util sort # ****************************************** SENTIEON_VERSION=$($SENTIEON_INSTALL_DIR/bin/sentieon driver --version) if (( $(echo "${SENTIEON_VERSION##*-} < 201911" |bc -l) )); then ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$RGID\tSM:$SM\tPL:$PL" -t $NT \ -K 100000000 -Y $FASTA $FASTQ_1 $FASTQ_2 || { echo -n 'bwa error'; exit 1; } ) | \ ( $SAMBLASTER --addMateTags -a || { echo -n 'samblaster error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -r $FASTA -o sorted.bam -t $NT --sam2bam -i - else #Sentieon 201911 and higher use BWA 0.7.17, which already produce MC tags in the output ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$RGID\tSM:$SM\tPL:$PL" -t $NT \ -K 100000000 -Y $FASTA $FASTQ_1 $FASTQ_2 || { echo -n 'error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -r $FASTA -o sorted.bam -t $NT --sam2bam -i - fi # ****************************************** # 2. Mark Duplicates with Sentieon # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i sorted.bam --algo LocusCollector \ --fun score_info score.txt || { echo "LocusCollector failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i sorted.bam --algo Dedup --score_info score.txt \ --metrics mark_dup_metrics.txt --output_dup_read_name tmp_dup_qname.txt || \ { echo "Dedup1 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i sorted.bam --algo Dedup \ --dup_read_name tmp_dup_qname.txt markduped.bam || { echo "Dedup2 failed"; exit 1; } # ****************************************** # 3. Base Quality Score Recalibration with Sentieon # ****************************************** interval_arg="--interval chr1,chr2,chr3,chr4,chr5,chr6,chr7,chr8,chr9,chr10,chr11,\ chr12,chr13,chr14,chr15,chr16,chr17,chr18,chr19,chr20,chr21,chr22" $SENTIEON_INSTALL_DIR/bin/sentieon driver $interval_arg -r $FASTA -t $NT -i markduped.bam \ --algo QualCal -k $KNOWN_MILLS -k $KNOWN_INDELS -k $KNOWN_DBSNP recal_data.table || \ { echo "QualCal failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i markduped.bam \ --read_filter QualCalFilter,table=recal_data.table,prior=-1.0,indel=false,levels=10/20/30,min_qual=6 \ --algo ReadWriter recaled_RW.cram || { echo "ReadWriter failed"; exit 1; } # ****************************************** # 4. Haplotyper with Sentieon # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i recaled_RW.cram --algo Haplotyper \ Haplotyper.vcf.gz || { echo "Haplotyper failed"; exit 1; } ``` ### 4.1.4 DNAseq® 多样本FASTQ分析脚本 以下是Sentieon DNAseq用于处理单个样本多组fastq文件的分析脚本示例: ``` #!/bin/sh # Copyright (c) 2016-2020 Sentieon Inc. All rights reserved # ******************************************* # Script to perform DNA seq variant calling # using a single sample with more than one # set of input fastq files (in this example # named set1_1.fastq.gz, set2_1.fastq.gz # set3_1.fastq.gz and set4_1.fastq.gz) # ******************************************* set -eu # Update with the fullpath location of your sample fastq SM="sample" #sample name RGID_PREFIX="rg_$SM" #read group ID PL="ILLUMINA" #or other sequencing platform FASTQ_FOLDER="/home/pipeline/samples" NUM_SETS="4" FASTQ_PREFIX="set" FASTQ_SUFFIX_1="_1.fastq.gz" FASTQ_SUFFIX_2="_2.fastq.gz" #If using Illumina paired data # Update with the location of the reference data files FASTA_DIR="/home/regression/references/b37/" FASTA="$FASTA_DIR/human_g1k_v37_decoy.fasta" KNOWN_DBSNP="$FASTA_DIR/dbsnp_138.b37.vcf.gz" KNOWN_INDELS="$FASTA_DIR/1000G_phase1.indels.b37.vcf.gz" KNOWN_MILLS="$FASTA_DIR/Mills_and_1000G_gold_standard.indels.b37.vcf.gz" # Update with the location of the Sentieon software package and license file SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-|release_version| export SENTIEON_LICENSE=/home/Licenses/Sentieon.lic #or using licsrvr: c1n11.sentieon.com:5443 # Other settings NT=$(nproc) #number of threads to use in computation, set to number of cores in the server START_DIR="$PWD/test/DNAseq_multiFASTQ" #Determine where the output files will be stored # You do not need to modify any of the lines below unless you want to tweak the pipeline # ************************************************************************************************************************************************************************ # ****************************************** # 0. Setup # ****************************************** WORKDIR="$START_DIR" mkdir -p $WORKDIR LOGFILE=$WORKDIR/run.log exec >$LOGFILE 2>&1 cd $WORKDIR # ****************************************** # 1. Mapping each set of input fastq with BWA-MEM, sorting # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 BAM_INPUT="" for i in $(seq 1 $NUM_SETS); do BAM_INPUT="$BAM_INPUT -i sorted_set$i.bam" ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem \ -R "@RG\tID:${RGID_PREFIX}_$i\tSM:$SM\tPL:$PL" -t $NT -K 10000000 $FASTA \ $FASTQ_FOLDER/$FASTQ_PREFIX$i$FASTQ_SUFFIX_1 \ $FASTQ_FOLDER/$FASTQ_PREFIX$i$FASTQ_SUFFIX_2 || { echo -n 'bwa error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -r $FASTA -o sorted_set$i.bam \ -t $NT --sam2bam -i - || { echo "Alignment failed"; exit 1; } done # ****************************************** # 2. Metrics on the multiple sorted BAM files # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT $BAM_INPUT \ --algo MeanQualityByCycle mq_metrics.txt --algo QualDistribution qd_metrics.txt \ --algo GCBias --summary gc_summary.txt gc_metrics.txt --algo AlignmentStat \ --adapter_seq '' aln_metrics.txt --algo InsertSizeMetricAlgo is_metrics.txt || \ { echo "Metrics failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o gc-report.pdf gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution -o qd-report.pdf qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle -o mq-report.pdf mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo -o is-report.pdf is_metrics.txt # ****************************************** # 3. Remove Duplicate Reads. It is possible # to remove instead of mark duplicates # by adding the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT $BAM_INPUT --algo LocusCollector \ --fun score_info score.txt || { echo "LocusCollector failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT $BAM_INPUT --algo Dedup \ --score_info score.txt --metrics dedup_metrics.txt deduped.bam || \ { echo "Dedup failed"; exit 1; } # ****************************************** # 2a. Coverage metrics # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i deduped.bam \ --algo CoverageMetrics coverage_metrics || { echo "CoverageMetrics failed"; exit 1; } # ****************************************** # 5. Base recalibration # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i deduped.bam --algo QualCal \ -k $KNOWN_DBSNP -k $KNOWN_MILLS -k $KNOWN_INDELS recal_data.table $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i deduped.bam -q recal_data.table \ --algo QualCal -k $KNOWN_DBSNP -k $KNOWN_MILLS -k $KNOWN_INDELS recal_data.table.post $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT --algo QualCal --plot \ --before recal_data.table --after recal_data.table.post recal.csv $SENTIEON_INSTALL_DIR/bin/sentieon plot QualCal -o recal_plots.pdf recal.csv # ****************************************** # 6b. HC Variant caller # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i deduped.bam -q recal_data.table \ --algo Haplotyper -d $KNOWN_DBSNP output-hc.vcf.gz || { echo "Haplotyper failed"; exit 1; } ``` ### 4.1.5 DNAseq® 全外显子测序分析脚本 以下是Sentieon DNAseq在全外显子测序(WES)中的分析脚本示例: ``` #!/bin/sh # Copyright (c) 2016-2020 Sentieon Inc. All rights reserved # ******************************************* # Script to perform DNA seq variant calling # using an exome sample with fastq files # named 1.fastq.gz and 2.fastq.gz # ******************************************* set -eu # Update with the fullpath location of your sample fastq SM="sample" #sample name RGID="rg_$SM" #read group ID PL="ILLUMINA" #or other sequencing platform FASTQ_FOLDER="/home/pipeline/samples" FASTQ_1="$FASTQ_FOLDER/1.fastq.gz" FASTQ_2="$FASTQ_FOLDER/2.fastq.gz" #If using Illumina paired data # Update with the location of the reference data files FASTA_DIR="/home/regression/references/b37/" FASTA="$FASTA_DIR/human_g1k_v37_decoy.fasta" KNOWN_DBSNP="$FASTA_DIR/dbsnp_138.b37.vcf.gz" KNOWN_INDELS="$FASTA_DIR/1000G_phase1.indels.b37.vcf.gz" KNOWN_MILLS="$FASTA_DIR/Mills_and_1000G_gold_standard.indels.b37.vcf.gz" INTERVAL_FILE="$FASTA_DIR/TruSeq_exome_targeted_regions.b37.bed" # Update with the location of the Sentieon software package and license file SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-|release_version| export SENTIEON_LICENSE=/home/Licenses/Sentieon.lic #or using licsrvr: c1n11.sentieon.com:5443 # Other settings NT=$(nproc) #number of threads to use in computation, set to number of cores in the server START_DIR="$PWD/test/DNAseq_interval" #Determine where the output files will be stored # You do not need to modify any of the lines below unless you want to tweak the pipeline # ************************************************************************************************************************************************************************ # ****************************************** # 0. Setup # ****************************************** WORKDIR="$START_DIR/${SM}" mkdir -p $WORKDIR LOGFILE=$WORKDIR/run.log exec >$LOGFILE 2>&1 cd $WORKDIR # ****************************************** # 1. Mapping reads with BWA-MEM, sorting # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$RGID\tSM:$SM\tPL:$PL" \ -t $NT -K 10000000 $FASTA $FASTQ_1 $FASTQ_2 || { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -r $FASTA -o sorted.bam -t $NT \ --sam2bam -i - || { echo "Alignment failed"; exit 1; } # ****************************************** # 2. Metrics # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT \ ${INTERVAL_FILE:+--interval $INTERVAL_FILE} -i sorted.bam --algo MeanQualityByCycle \ mq_metrics.txt --algo QualDistribution qd_metrics.txt --algo GCBias \ --summary gc_summary.txt gc_metrics.txt --algo AlignmentStat --adapter_seq '' \ aln_metrics.txt --algo InsertSizeMetricAlgo is_metrics.txt || \ { echo "Metrics failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o gc-report.pdf gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution -o qd-report.pdf qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle -o mq-report.pdf mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo -o is-report.pdf is_metrics.txt # ****************************************** # 3. Remove Duplicate Reads. It is possible # to remove instead of mark duplicates # by adding the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i sorted.bam --algo LocusCollector \ --fun score_info score.txt || { echo "LocusCollector failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i sorted.bam --algo Dedup \ --score_info score.txt --metrics dedup_metrics.txt deduped.bam || \ { echo "Dedup failed"; exit 1; } # ****************************************** # 2a. Coverage metrics # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT \ ${INTERVAL_FILE:+--interval $INTERVAL_FILE} -i deduped.bam --algo CoverageMetrics \ coverage_metrics || { echo "CoverageMetrics failed"; exit 1; } # ****************************************** # 5. Base recalibration # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT \ ${INTERVAL_FILE:+--interval $INTERVAL_FILE} -i deduped.bam --algo QualCal \ -k $KNOWN_DBSNP -k $KNOWN_MILLS -k $KNOWN_INDELS recal_data.table $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT \ ${INTERVAL_FILE:+--interval $INTERVAL_FILE} -i deduped.bam -q recal_data.table \ --algo QualCal -k $KNOWN_DBSNP -k $KNOWN_MILLS -k $KNOWN_INDELS recal_data.table.post $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT --algo QualCal --plot \ --before recal_data.table --after recal_data.table.post recal.csv $SENTIEON_INSTALL_DIR/bin/sentieon plot QualCal -o recal_plots.pdf recal.csv # ****************************************** # 6b. HC Variant caller # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA \ ${INTERVAL_FILE:+--interval $INTERVAL_FILE} -t $NT -i deduped.bam -q recal_data.table \ --algo Haplotyper -d $KNOWN_DBSNP --emit_conf=30 --call_conf=30 output-hc.vcf.gz || \ { echo "Haplotyper failed"; exit 1; } # ****************************************** # 5b. ReadWriter to output recalibrated bam # This stage is optional as variant callers # can perform the recalibration on the fly # using the before recalibration bam plus # the recalibration table # This stage should not include interval # option to prevent read loss # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i deduped.bam \ -q recal_data.table --algo ReadWriter recaled.bam || \ { echo "ReadWriter failed"; exit 1; } ``` ### 4.1.6 DNAseq® 全基因组测序分析脚本 以下是Sentieon DNAseq在全基因组测序(WGS)中的分析脚本示例: ``` #!/bin/sh # Copyright (c) 2016-2020 Sentieon Inc. All rights reserved # ******************************************* # Script to perform DNA seq variant calling # using a single sample with fastq files # named 1.fastq.gz and 2.fastq.gz # ******************************************* set -eu # Update with the fullpath location of your sample fastq SM="sample" #sample name RGID="rg_$SM" #read group ID PL="ILLUMINA" #or other sequencing platform FASTQ_FOLDER="/home/pipeline/samples" FASTQ_1="$FASTQ_FOLDER/1.fastq.gz" FASTQ_2="$FASTQ_FOLDER/2.fastq.gz" #If using Illumina paired data # Update with the location of the reference data files FASTA_DIR="/home/regression/references/b37/" FASTA="$FASTA_DIR/human_g1k_v37_decoy.fasta" KNOWN_DBSNP="$FASTA_DIR/dbsnp_138.b37.vcf.gz" KNOWN_INDELS="$FASTA_DIR/1000G_phase1.indels.b37.vcf.gz" KNOWN_MILLS="$FASTA_DIR/Mills_and_1000G_gold_standard.indels.b37.vcf.gz" # Update with the location of the Sentieon software package and license file SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-|release_version| export SENTIEON_LICENSE=/home/Licenses/Sentieon.lic #or using licsrvr: c1n11.sentieon.com:5443 # Other settings NT=$(nproc) #number of threads to use in computation, set to number of cores in the server START_DIR="$PWD/test/DNAseq" #Determine where the output files will be stored # You do not need to modify any of the lines below unless you want to tweak the pipeline # ************************************************************************************************************************************************************************ # ****************************************** # 0. Setup # ****************************************** WORKDIR="$START_DIR/${SM}" mkdir -p $WORKDIR LOGFILE=$WORKDIR/run.log exec >$LOGFILE 2>&1 cd $WORKDIR # ****************************************** # 1. Mapping reads with BWA-MEM, sorting # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$RGID\tSM:$SM\tPL:$PL" \ -t $NT -K 10000000 $FASTA $FASTQ_1 $FASTQ_2 || { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -r $FASTA -o sorted.bam -t $NT \ --sam2bam -i - || { echo "Alignment failed"; exit 1; } # ****************************************** # 2. Metrics # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i sorted.bam \ --algo MeanQualityByCycle mq_metrics.txt --algo QualDistribution qd_metrics.txt \ --algo GCBias --summary gc_summary.txt gc_metrics.txt --algo AlignmentStat \ --adapter_seq '' aln_metrics.txt --algo InsertSizeMetricAlgo is_metrics.txt || \ { echo "Metrics failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o gc-report.pdf gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution -o qd-report.pdf qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle -o mq-report.pdf mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo -o is-report.pdf is_metrics.txt # ****************************************** # 3. Remove Duplicate Reads. It is possible # to remove instead of mark duplicates # by adding the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i sorted.bam --algo LocusCollector \ --fun score_info score.txt || { echo "LocusCollector failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i sorted.bam --algo Dedup \ --score_info score.txt --metrics dedup_metrics.txt deduped.bam || \ { echo "Dedup failed"; exit 1; } # ****************************************** # 5. Base recalibration # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i deduped.bam --algo QualCal \ -k $KNOWN_DBSNP -k $KNOWN_MILLS -k $KNOWN_INDELS recal_data.table $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i deduped.bam -q recal_data.table \ --algo QualCal -k $KNOWN_DBSNP -k $KNOWN_MILLS -k $KNOWN_INDELS recal_data.table.post $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT --algo QualCal --plot \ --before recal_data.table --after recal_data.table.post recal.csv $SENTIEON_INSTALL_DIR/bin/sentieon plot QualCal -o recal_plots.pdf recal.csv # ****************************************** # 6b. HC Variant caller # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i deduped.bam -q recal_data.table \ --algo Haplotyper -d $KNOWN_DBSNP output-hc.vcf.gz || \ { echo "Haplotyper failed"; exit 1; } # ****************************************** # 5b. ReadWriter to output recalibrated bam # This stage is optional as variant callers # can perform the recalibration on the fly # using the before recalibration bam plus # the recalibration table # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i deduped.bam -q recal_data.table \ --algo ReadWriter recaled.bam || { echo "ReadWriter failed"; exit 1; } ``` ## 4.2 DNAscope Sentieon® Genomics软件包含一个改进的算法来执行胚系DNA分析的变异检测步骤。DNAscope使用的流程类似于DNAseq®中描述的流程,但在比对和变异检测阶段均存在差异。DNAscope支持使用模型文件以提高处理速度和准确性,除了检测SNP和短indel外,还可以执行结构变异检测。DNAscope推荐用于人类或其他哺乳动物样本的测序数据集。 ### 4.2.1 概述 在这个生信分析流程中,您需要以下输入文件: - FASTA 文件:包含与待分析样本对应的参考基因组核苷酸序列。 - FASTQ文件:一个或多个包含待分析样本核苷酸序列的文件。这些文件包含来自DNA测序的原始读数。软件支持输入使用GZIP压缩的FASTQ文件。软件仅支持包含Sanger格式(Phred+33)质量分数的文件。 - 机器学习模型文件:可从 https://github.com/Sentieon/sentieon-models 获取特定测序平台机器学习模型文件。 - (可选)包含变异检测区间的 BED 文件。推荐用于全外显子组或靶向测序数据。 - (可选)您希望包含在流程中的单核苷酸多态性数据库(dbSNP)数据。数据以 VCF 文件的形式使用;您可以使用经过 bgzip 压缩并建立索引的 VCF 文件。 DNAscope的典型生信分析流程包括以下步骤: 1. 将读数映射到参考基因组:此步骤将 FASTQ 文件中的读段比对并映射到 FASTA 文件中的参考基因组上。该步骤确保了数据能够被置于基因组上下文背景中(确定其位置)。 3. 计算数据指标:此步骤生成关于数据质量和流程分析质量的统计摘要。 4. 去除或标记重复序列:此步骤检测表明同一DNA分子被多次测序的读数。这些重复序列不具有信息性,不应作为额外的证据进行计数。 5. 使用 DNAscope 结合机器学习模型进行变异检测:此步骤识别数据中相对于参考基因组出现变异的位点,并计算每个样本在这些位点上的基因型。 ### 4.2.2 DNAscope使用步骤 #### (1)将读段映射到参考基因组 运行单条命令以高效地使用BWA执行比对,并使用Sentieon®软件创建BAM文件并进行排序: ``` (sentieon bwa mem -R '@RG\tID:GROUP_NAME\tSM:SAMPLE_NAME\tPL:PLATFORM' \ -t NUMBER_THREADS -x DNASCOPE_MODEL/bwa.model REFERENCE FASTQ [FASTQ2] \ || echo -n 'error' ) \ | sentieon util sort -r REFERENCE -o SORTED_BAM -t NUMBER_THREADS --sam2bam -i - ``` BWA的输入和选项在手册中有详细说明。 与DNAseq®中描述的BWA用法相比,此处通过参数 `-x DNASCOPE_MODEL/bwa.model` 增加了 `DNASCOPE_MODEL`(DNAscope 模型)。 该命令需要以下输入参数: - `GROUP_NAME`:将添加到读组头行的读组标识符。RG:ID在所有您计划使用的所有数据集中必须是唯一的,这在处理多个输入文件或执行肿瘤-正常对照分析时尤为重要。 - `SAMPLE_NAME`:将添加到读组头行的样本名称。 - `PLATFORM`:用于测序DNA的测序平台名称。可能的选项有:ILLUMINA(当fastq文件在Illumina™机器上产生时);IONTORRENT(当fastq文件在Life Technologies™ Ion-Torrent™机器上产生时);ELEMENT(当fastq文件在Element Biosciences™机器上产生时);DNBSEQ(当fastq文件在MGI™机器上产生时);ULTIMA(当fastq文件在Ultima Genomics™机器上产生时)。 - `NUMBER_THREADS`:计算中使用的线程数。我们建议该数量不要超过系统中可用的计算核心数。我们建议BWA和util二进制文件使用相同数量的线程。 - `REFERENCE`:参考FASTA文件的路径。您应确保*表4-1*中指定的所有额外参考数据都在同一位置,并具有一致的命名。 - `FASTQ`:样本FASTQ文件的路径。如果数据来自双端测序技术,您还需要输入FASTQ2作为相应的配对样本FASTQ文件。 - `SORTED_BAM`:映射并排序后的 BAM 输出文件的路径和文件名。系统将同时创建一个对应的索引文件(.bai)。 - `DNASCOPE_MODEL`:DNAscope模型包的路径。该模型将用于确定比对和变异检测阶段所使用的设置。 BWA会根据命令中使用的线程数产生略微不同的比对结果。这是因为 BWA 是基于数据块来计算插入片段大小分布的,而数据块的大小取决于线程数。为了保证结果与使用的线程数无关,您应该使用选项`-K 10000000`来固定数据块大小(以碱基为单位)。 对于指标收集和重复去除阶段,请参考 *第4.1.2节* 的获取详细的使用说明。 #### (2)使用机器学习模型进行胚系变异检测 建议使用带机器学习模型的DNAscope来执行变异检测,通过改进候选检测和过滤步骤,来实现更高的准确性。 Sentieon®可以为您提供特定测序平台的模型,该模型使用 https://github.com/genome-in-a-bottle 中GiAB真集的部分数据训练而成的。这些模型是通过将参考样本HG001-HG007进行处理而创建,流程包括 Sentieon® BWA-mem 比对和 Sentieon® 去重,并利用变异检测结果校准模型以拟合真集。 此外,Sentieon®可以协助您使用自有数据创建模型,从而针对您特定的测序和生物信息分析流程进行校准。 **1)在DNAscope中使用机器学习模型** 运行两条单独的命令来检测变异并应用机器学习模型。输入的BAM文件应该来自仅执行了比对和去重(没有BQSR或indel重新比对)的流程,以匹配模型创建方法。 ``` PCRFREE=true #PCRFREE=true means the sample is PCRFree, change it to false for PCR samples. if [ "$PCRFREE" = true ] ; then sentieon driver -t NUMBER_THREADS -r REFERENCE -i DEDUPED_BAM \ [--interval INTERVAL_FILE] --algo DNAscope [-d dbSNP] \ --pcr_indel_model none --model DNASCOPE_MODEL/dnascope.model \ TMP_VARIANT_VCF else sentieon driver -t NUMBER_THREADS -r REFERENCE -i DEDUPED_BAM \ [--interval INTERVAL_FILE] --algo DNAscope [-d dbSNP] \ --model DNASCOPE_MODEL/dnascope.model TMP_VARIANT_VCF fi sentieon driver -t NUMBER_THREADS -r REFERENCE --algo DNAModelApply \ --model DNASCOPE_MODEL/dnascope.model -v TMP_VARIANT_VCF VARIANT_VCF ``` **提醒:** 如果您使用的数据是不含PCR的,运行 DNAscope 时务必添加选项 `--pcr_indel_model NONE`。 根据测序是否涉及PCR步骤,DNAscope 在寻找显著的 INDEL 变异时会使用不同的先验概率,这可以通过 `--pcr_indel_model` 选项进行控制。默认的 `--pcr_indel_model` 设置针对的是 PCR 样本。因此,对于无 PCR 样本,设置 `--pcr_indel_model none` 至关重要。 该命令需要以下输入参数: - `NUMBER_THREADS`:计算中将使用的线程数。建议该数量不要超过系统中可用的计算核心数。 - `REFERENCE`:参考FASTA文件的路径。需确保参考序列与映射阶段使用的完全一致。 - `DEDUPED_BAM`:输入BAM文件的路径和文件名。 - `TMP_VARIANT_VCF`:DNAscope变异检测输出的路径和文件名。这是一个临时文件。 - `VARIANT_VCF`:变异检测输出的路径和文件名。系统将创建相应的索引文件。如果使用 `.gz` 扩展名,工具将输出压缩文件。 - `DNASCOPE_MODEL`:机器学习模型文件的路径。在DNAscope命令中,该模型将用于确定比对和变异检测阶段所使用的设置。 **提醒:** 使用机器学习模型运行DNAscope时,绝大多数高级设置均由模型本身决定;除 `--pcr_indel_model` 选项外,为其他选项设置特定值可能会对结果产生负面影响。 该命令的以下输入参数是可选的: - `INTERVAL_FILE`:BED文件的路径和文件名。 - `dbSNP`:将用于标记已知变异的单核苷酸多态性数据库(dbSNP)的路径和文件名。您只能使用一个dbSNP文件。 **2)使用DNAscope生成GVCF输出文件** 从 202112.04 版本开始,DNAscope 支持配合模型生成基因组 VCF格式的变异检测结果。GVCF 格式包含了被分析样本中参考等位基因纯合位点的额外信息。此功能需要使用近期训练的 DNAscope 模型;如果使用 202112.01 或更早版本的 Sentieon 训练的模型,则会导致运行报错。 运行两条单独的命令来检测变异并应用机器学习模型。输入的BAM文件应该来自仅执行了比对和去重的流程,以匹配模型的创建方法。 ``` PCRFREE=true #PCRFREE=true means the sample is PCRFree, change it to false for PCR samples. if [ "$PCRFREE" = true ] ; then sentieon driver -t NUMBER_THREADS -r REFERENCE -i DEDUPED_BAM \ [--interval INTERVAL_FILE] --algo DNAscope [-d dbSNP] \ --pcr_indel_model none --model DNASCOPE_MODEL/dnascope.model \ --emit_mode gvcf TMP_VARIANT_GVCF else sentieon driver -t NUMBER_THREADS -r REFERENCE -i DEDUPED_BAM \ [--interval INTERVAL_FILE] --algo DNAscope [-d dbSNP] \ --model DNASCOPE_MODEL/dnascope.model --emit_mode gvcf TMP_VARIANT_GVCF fi sentieon driver -t NUMBER_THREADS -r REFERENCE --algo DNAModelApply \ --model DNASCOPE_MODEL/dnascope.model -v TMP_VARIANT_GVCF VARIANT_GVCF ``` **提醒:** 如果您使用的数据是不含PCR的,在运行DNAscope时务必添加`--pcr_indel_model NONE`选项。 根据测序是否包含 PCR 步骤,DNAscope 在寻找显著的 INDEL 变异时会使用不同的先验概率 ,这可以通过 `--pcr_indel_model` 选项进行控制。默认的 `--pcr_indel_model` 设置针对的是 PCR 样本。因此,对于无 PCR 样本,设置 `--pcr_indel_model none` 至关重要。 该命令需要以下输入参数: - `NUMBER_THREADS`:计算中将使用的线程数。建议该数量不要超过系统中可用的计算核心数。 - `REFERENCE`:参考FASTA文件的路径。需确保参考序列与映射阶段使用的完全一致。 - `DEDUPED_BAM`:输入BAM文件的路径和文件名。 - `TMP_VARIANT_GVCF`:DNAscope 生成的 GVCF 输出的路径及文件名。这是一个临时文件。 - `VARIANT_GVCF`:GVCF输出的路径和文件名。系统将创建相应的索引文件。如果使用 `.gz` 扩展名,工具将输出压缩文件。 - `DNASCOPE_MODEL`:机器学习模型文件的路径。在DNAscope命令中,该模型将用于确定变异检测阶段所使用的设置。 该命令的以下输入参数是可选的: - `INTERVAL_FILE`:BED文件的路径和文件名。 - `dbSNP`:将用于标记已知变异的单核苷酸多态性数据库(dbSNP)的路径和文件名。您只能使用一个dbSNP文件。 **3)对DNAscope生成的GVCF文件进行基因型分析** 使用 Sentieon 202112.06 及更高版本中的 `GVCFtyper` 算法,可以对生成的 GVCF 输出文件进行单样本基因分型,或与其他样本的 GVCF 进行联合分型,最终输出单样本或多样本的 VCF 文件。 ``` sentieon driver -r REFERENCE --algo GVCFtyper \ -v s1_VARIANT_GVCF -v s2_VARIANT_GVCF -v s3_VARIANT_GVCF VARIANT_VCF ``` 请查看Sentieon手册以获取有关GVCFtyper算法的更多详细信息,https://support.sentieon.com/manual/usages/general/#gvcftyper-algorithm 。 **提醒:** GVCFtyper可用于将来自多个测序平台的DNAscope GVCF基因型分析为单个多样本VCF。 GVCFtyper不支持将 DNAscope GVCF 与未使用机器学习模型生成的 DNAscope GVCF 混合进行联合分型,也不支持将其与其他工具生成的 GVCF 混合处理。 #### (3)结构变异检测 为了执行结构变异检测,您需要在 DNAscope 命令中添加输出断裂端信息的选项;这可以通过启用 `bnd` 变异类型来实现。结构变异检测由两条独立的命令完成。 ``` sentieon driver -t NUMBER_THREADS -r REFERENCE -i DEDUPED_BAM \ --algo DNAscope --var_type bnd \ [-d dbSNP] TMP_VARIANT_VCF sentieon driver -t NUMBER_THREADS -r REFERENCE --algo SVSolver \ -v TMP_VARIANT_VCF STRUCTURAL_VARIANT_VCF ``` 该命令需要以下输入参数: - `NUMBER_THREADS`:计算中将使用的线程数。我们建议该数量不要超过系统中可用的计算核心数。 - `REFERENCE`:参考FASTA文件的路径及文件名。需确保参考序列与映射阶段使用的完全一致。 - `DEDUPED_BAM`:输入BAM文件的路径和文件名。 - `TMP_VARIANT_VCF`:DNAscope 生成的包含 BND 信息的变异检测输出文件的路径及文件名。在检测结构变异时,这是一个临时文件。 - `STRUCTURAL_VARIANT_VCF`:包含结构变异检测结果的输出文件的路径及文件名。系统将创建相应的索引文件。如果使用 `.gz` 扩展名,工具将输出压缩文件。 该命令的以下输入参数是可选的: - `dbSNP`:将用于标记已知变异的单核苷酸多态性数据库(dbSNP)的路径和文件名。您只能使用一个dbSNP文件。 **请注意**,结构变异检测与DNAscope模型输入文件不兼容。特别是,当设置了`--var_type BND`时,应避免使用`--model`选项。 ### 4.2.3 DNAscope 全基因组测序分析脚本 以下是Sentieon DNAscope在全基因组测序(WGS)中的分析脚本示例: ``` #!/bin/sh # Copyright (c) 2016-2023 Sentieon Inc. All rights reserved # ******************************************* # Script to perform DNAscope variant calling # using a single sample with fastq files # named 1.fastq.gz and 2.fastq.gz # ******************************************* set -eu # Update with the fullpath location of your sample fastq SM="sample" #sample name RGID="rg_$SM" #read group ID PL="ILLUMINA" #or other sequencing platform FASTQ_FOLDER="/home/pipeline/samples" FASTQ_1="$FASTQ_FOLDER/1.fastq.gz" FASTQ_2="$FASTQ_FOLDER/2.fastq.gz" #If using Illumina paired data # Update with the location to the DNAscope model file # Model files can be found at, https://github.com/Sentieon/sentieon-models DNASCOPE_MODEL=/home/pipeline/models/DNAscopeIlluminaWGS2.0.bundle # Update with the location of the reference data files FASTA_DIR="/home/regression/references/b37/" FASTA="$FASTA_DIR/human_g1k_v37_decoy.fasta" KNOWN_DBSNP="$FASTA_DIR/dbsnp_138.b37.vcf.gz" KNOWN_INDELS="$FASTA_DIR/1000G_phase1.indels.b37.vcf.gz" KNOWN_MILLS="$FASTA_DIR/Mills_and_1000G_gold_standard.indels.b37.vcf.gz" # Update with the location of the Sentieon software package and license file SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-|release_version| export SENTIEON_LICENSE=/home/Licenses/Sentieon.lic #or using licsrvr: c1n11.sentieon.com:5443 # Other settings PCRFREE=true # The data was sequenced with a PCR-free library prep NT=$(nproc) #number of threads to use in computation, set to number of cores in the server START_DIR="$PWD/test/DNAscope" #Determine where the output files will be stored # You do not need to modify any of the lines below unless you want to tweak the pipeline # ************************************************************************************************************************************************************************ # ****************************************** # 0. Setup # ****************************************** WORKDIR="$START_DIR/${SM}" mkdir -p $WORKDIR LOGFILE=$WORKDIR/run.log exec >$LOGFILE 2>&1 cd $WORKDIR # ****************************************** # 1. Mapping reads with BWA-MEM, sorting # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$RGID\tSM:$SM\tPL:$PL" \ -t $NT -K 10000000 -x $DNASCOPE_MODEL/bwa.model \ $FASTA $FASTQ_1 $FASTQ_2 || { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -r $FASTA -o sorted.bam -t $NT \ --sam2bam -i - || { echo "Alignment failed"; exit 1; } # ****************************************** # 2. Metrics # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i sorted.bam \ --algo MeanQualityByCycle mq_metrics.txt --algo QualDistribution qd_metrics.txt \ --algo GCBias --summary gc_summary.txt gc_metrics.txt --algo AlignmentStat \ --adapter_seq '' aln_metrics.txt --algo InsertSizeMetricAlgo is_metrics.txt || \ { echo "Metrics failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o gc-report.pdf gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution -o qd-report.pdf qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle -o mq-report.pdf mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo -o is-report.pdf is_metrics.txt # ****************************************** # 3. Remove Duplicate Reads. It is possible # to remove instead of mark duplicates # by adding the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i sorted.bam --algo LocusCollector \ --fun score_info score.txt || { echo "LocusCollector failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i sorted.bam --algo Dedup \ --score_info score.txt --metrics dedup_metrics.txt deduped.bam || \ { echo "Dedup failed"; exit 1; } # ****************************************** # 4a. DNAscope variant calling # ****************************************** indel_model_arg="" if [ "$PCRFREE" = true ]; then indel_model_arg="--pcr_indel_model none" fi $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i deduped.bam \ --algo DNAscope $indel_model_arg --model $DNASCOPE_MODEL/dnascope.model \ -d $KNOWN_DBSNP output-ds_tmp.vcf.gz || \ { echo "DNAscope failed"; exit 1; } # ****************************************** # 4b. Variant filtering and genotyping # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT \ --algo DNAModelApply --model $DNASCOPE_MODEL/dnascope.model \ -v output-ds_tmp.vcf.gz output-ds.vcf.gz || \ { echo "DNAModelApply failed"; exit 1; } ``` ### 4.2.4 DNAscope 外显子测序分析脚本 以下是Sentieon DNAscope在全外显子测序(WES)中的分析脚本示例: ``` #!/bin/sh # Copyright (c) 2016-2023 Sentieon Inc. All rights reserved # ******************************************* # Script to perform DNAscope variant calling # using an exome sample with fastq files # named 1.fastq.gz and 2.fastq.gz # ******************************************* set -eu # Update with the fullpath location of your sample fastq SM="sample" #sample name RGID="rg_$SM" #read group ID PL="ILLUMINA" #or other sequencing platform FASTQ_FOLDER="/home/pipeline/samples" FASTQ_1="$FASTQ_FOLDER/1.fastq.gz" FASTQ_2="$FASTQ_FOLDER/2.fastq.gz" #If using Illumina paired data # Update with the location to the DNAscope model file # Model files can be found at, https://github.com/Sentieon/sentieon-models DNASCOPE_MODEL=/home/pipeline/models/DNAscopeIlluminaWES2.0.bundle # Update with the location of the reference data files FASTA_DIR="/home/regression/references/b37/" FASTA="$FASTA_DIR/human_g1k_v37_decoy.fasta" KNOWN_DBSNP="$FASTA_DIR/dbsnp_138.b37.vcf.gz" KNOWN_INDELS="$FASTA_DIR/1000G_phase1.indels.b37.vcf.gz" KNOWN_MILLS="$FASTA_DIR/Mills_and_1000G_gold_standard.indels.b37.vcf.gz" INTERVAL_FILE="$FASTA_DIR/TruSeq_exome_targeted_regions.b37.bed" # Update with the location of the Sentieon software package and license file SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-|release_version| export SENTIEON_LICENSE=/home/Licenses/Sentieon.lic #or using licsrvr: c1n11.sentieon.com:5443 # Other settings PCRFREE=true # The data was sequenced with a PCR-free library prep NT=$(nproc) #number of threads to use in computation, set to number of cores in the server START_DIR="$PWD/test/DNAscope" #Determine where the output files will be stored # You do not need to modify any of the lines below unless you want to tweak the pipeline # ************************************************************************************************************************************************************************ # ****************************************** # 0. Setup # ****************************************** WORKDIR="$START_DIR/${SM}" mkdir -p $WORKDIR LOGFILE=$WORKDIR/run.log exec >$LOGFILE 2>&1 cd $WORKDIR # ****************************************** # 1. Mapping reads with BWA-MEM, sorting # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$RGID\tSM:$SM\tPL:$PL" \ -t $NT -K 10000000 -x $DNASCOPE_MODEL/bwa.model \ $FASTA $FASTQ_1 $FASTQ_2 || { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -r $FASTA -o sorted.bam -t $NT \ --sam2bam -i - || { echo "Alignment failed"; exit 1; } # ****************************************** # 2. Metrics # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT \ ${INTERVAL_FILE:+--interval $INTERVAL_FILE} -i sorted.bam --algo MeanQualityByCycle \ mq_metrics.txt --algo QualDistribution qd_metrics.txt --algo GCBias \ --summary gc_summary.txt gc_metrics.txt --algo AlignmentStat --adapter_seq '' \ aln_metrics.txt --algo InsertSizeMetricAlgo is_metrics.txt || \ { echo "Metrics failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o gc-report.pdf gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution -o qd-report.pdf qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle -o mq-report.pdf mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo -o is-report.pdf is_metrics.txt # ****************************************** # 3. Remove Duplicate Reads. It is possible # to remove instead of mark duplicates # by adding the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i sorted.bam --algo LocusCollector \ --fun score_info score.txt || { echo "LocusCollector failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i sorted.bam --algo Dedup \ --score_info score.txt --metrics dedup_metrics.txt deduped.bam || \ { echo "Dedup failed"; exit 1; } # ****************************************** # 4a. DNAscope variant calling # ****************************************** indel_model_arg="" if [ "$PCRFREE" = true ]; then indel_model_arg="--pcr_indel_model none" fi $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i deduped.bam \ ${INTERVAL_FILE:+--interval $INTERVAL_FILE} --algo DNAscope $indel_model_arg \ --model $DNASCOPE_MODEL/dnascope.model -d $KNOWN_DBSNP output-ds_tmp.vcf.gz || \ { echo "DNAscope failed"; exit 1; } # ****************************************** # 4b. Variant filtering and genotyping # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT \ --algo DNAModelApply --model $DNASCOPE_MODEL/dnascope.model \ -v output-ds_tmp.vcf.gz output-ds.vcf.gz || \ { echo "DNAModelApply failed"; exit 1; } ``` ## 4.3 泛基因组流程 与 DNAseq® 或 DNAscope 流程中典型的线性比对相比,Sentieon 泛基因组流程利用泛基因组图数据结构来提高短读段的比对效率及变异检测的准确性。该泛基因组流程推荐用于人类样本的测序数据集。 ### 4.3.1 概述 #### (1)流程简介 Sentieon® 泛基因组流程将通过以下步骤处理输入的读段: 1. 提取 k-mer 谱:此步骤从输入的读段中提取 k-mer 特征,并生成一个 KFF 格式的文件。 2. 生成个性化泛基因组:利用样本的 k-mer 谱生成个性化泛基因组,随后将其转换为个性化参考基因组。 3. 将读段映射至参考基因组:此步骤将 FASTQ 文件中的读段比对到 FASTA 格式的参考基因组上。如果输入数据是已比对的 BAM 或 CRAM 格式,则跳过此步。 4. 将读段映射至个性化参考基因组:此步骤将读段的子集比对到样本的个性化参考基因组上,并将这些比对结果坐标回映射至标准参考基因组。 5. 重复序列标记与指标收集:此步骤识别被多次测序的 DNA 分子,并将这些读段标记为重复序列,以便在下游分析中将其排除。同时,该步骤会根据比对后的读段数据生成数据质量的统计摘要。如果系统中安装了 MultiQC,则会根据收集到的指标生成一份报告。标记重复序列后的 读段将以 BAM 或 CRAM 格式输出。 6. 使用 DNAscope 进行小变异检测:此步骤识别样本相对于参考基因组的小变异(包括 SNV 和 Indel),并计算样本的基因型。 上述步骤可以通过 `sentieon-cli` 工具以单条命令运行。 #### (2)输入文件 Sentieon® 泛基因组流程集成在 `sentieon-cli` 中。有关 `sentieon-cli` 的安装说明可以在 *4.8.1 Sentieon CLI* 章节中查找。关于该流程的详细描述可查阅 *4.8.5 Sentieon Pangenom* 章节。在该生信分析流程中,您需要准备以下输入: * 包含与待分析样本对应的 GRCh38 参考基因组核苷酸序列的 FASTA 文件。同时需要该参考基因组的 Samtools 索引文件和 BWA 索引文件。 * GBZ 格式的泛基因组文件及其对应的单倍型(.hapl)文件。该流程要求泛基因组中包含 GRCh38 的 contigs(重叠群)作为起始图结构。 * 一个或多个经过 GZIP 压缩的 FASTQ 文件,包含待分析样本的核苷酸序列。这些文件包含了 DNA 测序的原始读段。软件仅支持包含 Sanger 格式(Phred+33)质量分数的文件。 * 包含群体数据库等位基因频率信息的群体 VCF 文件。 * 特定于流程和测序平台的模型包文件。模型包文件可以从 [https://github.com/Sentieon/sentieon-models](https://github.com/Sentieon/sentieon-models) 获取。 * (可选)包含变异检测区间的 BED 文件。对于全基因组测序数据,建议使用该文件将变异检测限制在标准 contigs 内;对于全外显子组测序数据,建议以此将变异检测限制在目标区域内。 * (可选)您希望包含在流程中的单核苷酸多态性数据库(dbSNP)数据。该数据以 VCF 格式使用;您可以使用经过 bgzip 压缩并建立索引的 VCF 文件。 #### (3)必备工具 除了 Sentieon® 软件外,该生信分析流程还需要以下工具: * Samtools:1.16 或更高版本。用于对比对后的读段数据进行操作。 * Bcftools:1.22 或更高版本。用于对检测到的变异进行操作。 * vg:用于生成个性化泛基因组。 * KMC:第 3 版或更高版本。用于对输入样本进行 k-mer 计数。 以下工具为可选工具,可为流程提供额外功能: * MultiQC:用于根据流程产出的指标生成综合报告。 这些工具将直接从用户的环境变量 `PATH` 中调用。 ### 4.3.2 示例用法 以下示例展示了使用推荐的输入文件运行 Sentieon® 泛基因组流程处理测试样本的详细步骤。 #### (1)下载输入文件 运行以下 Shell 命令下载流程所需的 GRCh38 参考基因组、泛基因组及群体 VCF 输入文件: ``` curl-L \ -O 'https://ftp.sentieon.com/public/GRCh38/hg38_canonical.bed' \ -O 'https://human-pangenomics.s3.amazonaws.com/pangenomes/freeze/release2/minigraph-cactus/ hprc-v2.0-mc-grch38.gbz' \ -O 'https://human-pangenomics.s3.amazonaws.com/pangenomes/freeze/release2/minigraph-cactus/ → hprc-v2.0-mc-grch38.hapl' \ -O 'https://ftp.sentieon.com/public/GRCh38/population/pop-v20g41-20251216.vcf.gz' \ -o 'hg38_ucsc.fa' 'https://ngi-igenomes.s3.amazonaws.com/igenomes/Homo_sapiens/UCSC/hg38/ → Sequence/WholeGenomeFasta/genome.fa' \ -o 'hg38_ucsc.fa.fai' 'https://ngi-igenomes.s3.amazonaws.com/igenomes/Homo_sapiens/UCSC/ → hg38/Sequence/WholeGenomeFasta/genome.fa.fai' \ -o 'hg38_ucsc.fa.amb' 'https://ngi-igenomes.s3.amazonaws.com/igenomes/Homo_sapiens/UCSC/ → hg38/Sequence/BWAIndex/genome.fa.amb' \ -o 'hg38_ucsc.fa.ann' 'https://ngi-igenomes.s3.amazonaws.com/igenomes/Homo_sapiens/UCSC/ →hg38/Sequence/BWAIndex/genome.fa.ann' \ -o 'hg38_ucsc.fa.bwt' 'https://ngi-igenomes.s3.amazonaws.com/igenomes/Homo_sapiens/UCSC/ →hg38/Sequence/BWAIndex/genome.fa.bwt' \ -o 'hg38_ucsc.fa.pac' 'https://ngi-igenomes.s3.amazonaws.com/igenomes/Homo_sapiens/UCSC/ →hg38/Sequence/BWAIndex/genome.fa.pac' \ -o 'hg38_ucsc.fa.sa' 'https://ngi-igenomes.s3.amazonaws.com/igenomes/Homo_sapiens/UCSC/ →hg38/Sequence/BWAIndex/genome.fa.sa' \ -O 'https://storage.googleapis.com/gcp-public-data--broad-references/hg38/v0/Homo_sapiens_ →assembly38.dbsnp138.vcf.gz' \ -O 'https://storage.googleapis.com/gcp-public-data--broad-references/hg38/v0/Homo_sapiens_ →assembly38.dbsnp138.vcf.gz.tbi' ``` 该流程的模型包文件可以从 Sentieon 模型页面下载。 #### (2)使用 FASTQ 输入运行泛基因组流程 只需一条命令即可根据输入文件执行泛基因组分析流程: ``` sentieon-cli sentieon-pangenome \ -r hg38_ucsc.fa \ --hapl hprc-v2.0-mc-grch38.hapl \ --gbz hprc-v2.0-mc-grch38.gbz \ -m SentieonIlluminaPangenomeWGS2.0.bundle \ --pop_vcf pop-v20g41-20251216.vcf.gz \ --r1_fastq HG002.novaseq.pcr-free.30x.R1.fastq.gz \ --r2_fastq HG002.novaseq.pcr-free.30x.R2.fastq.gz \ --readgroup "@RG\tID:HG002-1\tSM:HG002\tLB:HG002-LB-1\tPL:ILLUMINA" \ -b hg38_canonical.bed \ --dbsnp Homo_sapiens_assembly38.dbsnp138.vcf.gz \ --pcr_free \ HG002_pangenome.vcf.gz ``` 根据测序是否包含 PCR 扩增步骤,DNAscope 在寻找显著的 INDEL 变异时会使用不同的先验概率。默认设置适用于标准建库的测序样本。设置 `--pcr_free` 参数将使用适用于无 PCR 建库测序样本的先验概率。 #### (3)输出文件 当启用所有可选功能时,流程将输出以下文件: * HG002_pangenome.vcf.gz:VCF 格式的 SNV 和 Indel 检测结果。 * HG002_pangenome_bwa_deduped.cram:基于输入 FASTQ 文件,经过 BWA 比对、坐标排序及重复序列标记后的 CRAM 数据。 * HG002_pangenome_mm2_deduped.cram:基于输入 FASTQ 文件,经过泛基因组比对、坐标排序及重复序列标记后的 CRAM 数据。此文件中的读段已比对至泛基因组并回映射至 GRCh38。 * HG002_pangenome_metrics/:包含被分析样本质控指标的目录。 ### 4.3.3 Sentieon® 泛基因组流程的限制 Sentieon® 泛基因组流程目前仅支持基于 GRCh38 参考序列的 Minigraph-Cactus 泛基因组,例如由人类泛基因组参考联盟(HPRC)生成的泛基因组。如需了解如何在其他泛基因组上使用该流程,请联系 Sentieon® 技术支持。 ## 4.4 TNseq® Sentieon® Genomics 软件的另一个典型用途是执行 Broad 研究所在“体细胞变异检测(SNVs + Indels)”中推荐的肿瘤-正常对照分析分析流程。图4-2说明了这样一个典型的生信分析流程。 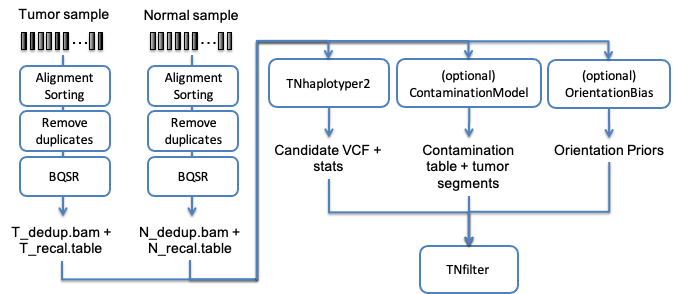 <center>图4-2 肿瘤-正常对照分析推荐的生信分析流程</center> <br> 请查看https://support.sentieon.com/appnotes/somatic/ 中的应用说明,了解不同输入的推荐体细胞变异检测流程。 ### 4.4.1 概述 在这个生物信息学流程中,您需要以下输入: - FASTA 文件:包含与待分析样本对应的参考基因组核苷酸序列。 - 两组 FASTQ 文件:包含待分析样本的核苷酸序列,一组用于肿瘤样本,一组用于匹配的正常样本。这些文件包含了 DNA 测序的原始读段。软件支持输入经过 GZIP 压缩的 FASTQ 文件。软件仅支持包含 Sanger 格式(Phred+33)质量分数的文件。 您还可以在流程中包含以下可选输入,这将帮助算法检测测序伪影并去除假阳性: - 正常样本面板VCF:包含从多个无关的正常样本中提取的、表现为变异的常见错误列表。该文件内容将用于识别更有可能是胚系变异的位点,并对其进行过滤。 - 人群资源VCF:包含群体特定等位基因频率的列表,用于过滤可能的胚系变异并对结果进行注释。 肿瘤-正常配对的典型生信分析流程包括以下步骤: 1. 使用类似 *第4.1.2节* 介绍的DNAseq流程独立预处理肿瘤和正常样本,包括以下阶段: - 将读段映射到参考基因组:您需要确保肿瘤和正常样本的SM样本标签不同,因为在体细胞变异检测中需要将其作为参数。 - 计算数据指标。 - 移除重复序列。 - (可选)碱基质量分数重校准(BQSR)。 2. 体细胞变异发现,包括以下阶段: - (可选)评估跨样本间污染和肿瘤分段。 - (可选)评估测序中可能存在的任何方向偏差。 - 对两个单独的BAM文件进行体细胞候选变异检测:此步骤识别癌症基因组数据相对于正常基因组显示体细胞变异的潜在位点,并计算该位点的基因型。 - 过滤变异。 ### 4.4.2 TNseq®使用步骤 对于映射、重复去除和碱基质量分数重校准阶段,请参考 *第4.1.2节* 获取详细的使用说明。 #### (1)使用配对正常样本进行变异检测 运行两条命令对肿瘤-正常配对进行变异检测。我们建议将可选步骤(估计跨样本污染和估计方向偏差)与`TNhaplotyper`在同一命令中运行,以提高性能。 ``` sentieon driver -t NUMBER_THREADS -r REFERENCE \ -i TUMOR_DEDUPED_BAM [-q TUMOR_RECAL_DATA.TABLE] \ -i NORMAL_DEDUPED_BAM [-q NORMAL_RECAL_DATA.TABLE] \ --algo TNhaplotyper2 --tumor_sample TUMOR_SAMPLE_NAME \ --normal_sample NORMAL_SAMPLE_NAME \ [--germline_vcf GERMLINE_RESOURCE] \ [--pon PANEL_OF_NORMAL] \ TMP_OUT_TN_VCF \ [ --algo OrientationBias --tumor_sample TUMOR_SAMPLE_NAME \ ORIENTATION_DATA ] \ [ --algo ContaminationModel --tumor_sample TUMOR_SAMPLE_NAME \ --normal_sample NORMAL_SAMPLE_NAME \ --vcf GERMLINE_RESOURCE \ --tumor_segments CONTAMINATION_DATA.segments \ CONTAMINATION_DATA ] sentieon driver -r REFERENCE \ --algo TNfilter --tumor_sample TUMOR_SAMPLE_NAME \ --normal_sample NORMAL_SAMPLE_NAME \ -v TMP_OUT_TN_VCF \ [--contamination CONTAMINATION_DATA] \ [--tumor_segments CONTAMINATION_DATA.segments] \ [--orientation_priors ORIENTATION_DATA] \ OUT_TN_VCF ``` 该命令需要以下输入参数: - `NUMBER_THREADS`:计算中将使用的线程数。建议该数量不要超过系统中可用的计算核心数。 - `REFERENCE`:参考FASTA文件的路径。需确保参考序列与映射阶段使用的完全一致。 - `TUMOR_DEDUPED_BAM`:肿瘤样本经过预处理(去重后)的 BAM 文件路径。 - `NORMAL_DEDUPED_BAM`:正常样本经过预处理(去重后)的 BAM 文件路径。 - `TUMOR_SAMPLE_NAME`:在映射阶段为肿瘤样本设置的样本名称。 - `NORMAL_SAMPLE_NAME`:在映射阶段为正常样本设置的样本名称。 - `TMP_OUT_TN_VCF`:TNhaplotyper2输出文件的路径和文件名;这是一个临时文件。 - `OUT_TN_VCF`:包含变异检测的输出文件的路径和文件名。 该命令的以下输入参数是可选的: - `TUMOR_RECAL_DATA.TABLE`:肿瘤样本BQSR阶段存储结果的路径。 - `NORMAL_RECAL_DATA.TABLE`:正常样本BQSR阶段存储结果的路径。 - `PANEL_OF_NORMAL`:正常样本面板VCF文件的路径和文件名。 - `GERMLINE_RESOURCE`:人群种系资源的路径。资源文件中的AF INFO字段将用于注释变异等位基因的人群等位基因频率,然后用于识别可能不是真正体细胞变异的种系变异。 - `ORIENTATION_DATA`:由OrientationBias生成的包含方向偏差信息的文件的路径和文件名。 - `CONTAMINATION_DATA`:由ContaminationModel生成的包含污染信息的文件的路径和文件名。 - `CONTAMINATION_DATA.segments`:由ContaminationModel生成的包含肿瘤分段信息的文件的路径和文件名。 #### (2)缺少配对正常样本时的特殊考虑 当缺少配对正常样本时,我们建议对上一节所示的流程进行以下调整: - 应包括`PANEL_OF_NORMA`L和`GERMLINE_RESOURCE`,因为它们替代了缺失的配对正常样本。同时缺少配对正常样本和这些种系资源将导致最终输出VCF中出现大量种系假阳性检测。 - 不应包括`--normal_sample`参数以及正常样本的输入BAM文件和重校准表。 #### (3)生成GERMLINE_RESOURCE种系人群资源 包含群体等位基因频率的种系人群资源文件可以通过以下程序从gnomAD获取并进行后处理: - 下载您所需版本的所有每条染色体`.vcf.bgz`文件。 - 对于每条染色体,移除不必要的注释以减小文件大小: ``` bcftools annotate -x ^INFO/AF,INFO/AC INPUT.chrN.vcf.bgz | bcftools norm -m +any -Oz -o OUTPUT.chrN.vcf.gz ``` - 将所有文件合并为一个文件,并创建相应的索引: ``` bcftools concat -Oz -o OUTPUT.vcf.gz OUTPUT.chr*.vcf.gz && bcftools index -t OUTPUT.vcf.gz ``` 例如,要从Google下载GnomAD v3.1版本,您可以运行: ``` for chr in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X Y; do curl https://storage.googleapis.com/gcp-public-data--gnomad/release/3.1/vcf/genomes/gnomad.genomes.v3.1.sites.chr$chr.vcf.bgz \ | bcftools annotate -x ^INFO/AF,INFO/AC - | bcftools norm -m +any -Oz -o tmp_OUTPUT.chr$chr.vcf.gz file_list="$file_list tmp_OUTPUT.chr$chr.vcf.gz" done bcftools concat -Oz -o OUTPUT.vcf.gz $file_list && bcftools index -t OUTPUT.vcf.gz rm $file_list ``` ### 4.4.3 TNseq® 配对样本全基因组分析脚本 以下是Sentieon TNseq对肿瘤-正常样本分析的全基因组脚本示例: ``` #!/bin/sh # Copyright (c) 2016-2024 Sentieon Inc. All rights reserved # ******************************************* # Script to perform TN seq variant calling # using a matched paired Tumor+normal sample with fastq # files named normal_1.fastq.gz, normal_2.fastq.gz # tumor_1.fastq.gz, tumor_2.fastq.gz # ******************************************* set -eu # Update with the fullpath location of your sample fastq TUMOR_SM="tumor_sample" #sample name TUMOR_RGID="rg_$TUMOR_SM" #read group ID NORMAL_SM="normal_sample" #sample name NORMAL_RGID="rg_$NORMAL_SM" #read group ID PL="ILLUMINA" #or other sequencing platform FASTQ_FOLDER="/home/pipeline/samples" TUMOR_FASTQ_1="$FASTQ_FOLDER/tumor_1.fastq.gz" TUMOR_FASTQ_2="$FASTQ_FOLDER/tumor_2.fastq.gz" #If using Illumina paired data NORMAL_FASTQ_1="$FASTQ_FOLDER/normal_1.fastq.gz" NORMAL_FASTQ_2="$FASTQ_FOLDER/normal_2.fastq.gz" # Update with the location of the reference data files FASTA_DIR="/home/regression/references/b37/" FASTA="$FASTA_DIR/human_g1k_v37_decoy.fasta" KNOWN_DBSNP="$FASTA_DIR/dbsnp_138.b37.vcf.gz" KNOWN_INDELS="$FASTA_DIR/1000G_phase1.indels.b37.vcf.gz" KNOWN_MILLS="$FASTA_DIR/Mills_and_1000G_gold_standard.indels.b37.vcf.gz" CONTAMINATION_VCF="$FASTA_DIR/germline_vcf-af-only-gnomad.raw.sites.vcf" # A VCF of germline sites to use for contamination detection PON= # the Mutect2 panel-of-normals VCF file GERMLINE_VCF= # A VCF of known germline sites # Update with the location of the Sentieon software package and license file SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-|release_version| export SENTIEON_LICENSE=/home/Licenses/Sentieon.lic #or using licsrvr: c1n11.sentieon.com:5443 # Other settings NT=$(nproc) #number of threads to use in computation, set to number of cores in the server START_DIR="$PWD/test/TNseq" #Determine where the output files will be stored # You do not need to modify any of the lines below unless you want to tweak the pipeline # ************************************************************************************************************************************************************************ # ****************************************** # 0. Setup # ****************************************** WORKDIR="$START_DIR" mkdir -p $WORKDIR LOGFILE=$WORKDIR/run.log exec >$LOGFILE 2>&1 cd $WORKDIR # ****************************************** # 1a. Mapping reads with BWA-MEM, sorting for tumor sample # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$TUMOR_RGID\tSM:$TUMOR_SM\tPL:$PL" \ -t $NT -K 10000000 $FASTA $TUMOR_FASTQ_1 $TUMOR_FASTQ_2 || \ { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -o tumor_sorted.bam -t $NT --sam2bam -i - || \ { echo "Alignment1 failed"; exit 1; } # ****************************************** # 1b. Mapping reads with BWA-MEM, sorting for normal sample # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$NORMAL_RGID\tSM:$NORMAL_SM\tPL:$PL" \ -t $NT -K 10000000 $FASTA $NORMAL_FASTQ_1 $NORMAL_FASTQ_2 || \ { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -o normal_sorted.bam -t $NT --sam2bam -i - || \ { echo "Alignment2 failed"; exit 1; } # ****************************************** # 2a. Metrics for tumor sample # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_sorted.bam \ --algo MeanQualityByCycle tumor_mq_metrics.txt \ --algo QualDistribution tumor_qd_metrics.txt --algo GCBias \ --summary tumor_gc_summary.txt tumor_gc_metrics.txt --algo AlignmentStat \ --adapter_seq '' tumor_aln_metrics.txt \ --algo InsertSizeMetricAlgo tumor_is_metrics.txt \ --algo CoverageMetrics --omit_base_output tumor_coverage_metrics || \ { echo "Metrics1 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o tumor_gc-report.pdf tumor_gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution \ -o tumor_qd-report.pdf tumor_qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle \ -o tumor_mq-report.pdf tumor_mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo \ -o tumor_is-report.pdf tumor_is_metrics.txt # ****************************************** # 2b. Metrics for normal sample # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i normal_sorted.bam \ --algo MeanQualityByCycle normal_mq_metrics.txt \ --algo QualDistribution normal_qd_metrics.txt --algo GCBias \ --summary normal_gc_summary.txt normal_gc_metrics.txt --algo AlignmentStat \ --adapter_seq '' normal_aln_metrics.txt \ --algo InsertSizeMetricAlgo normal_is_metrics.txt \ --algo CoverageMetrics --omit_base_output normal_coverage_metrics || \ { echo "Metrics2 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o normal_gc-report.pdf normal_gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution \ -o normal_qd-report.pdf normal_qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle \ -o normal_mq-report.pdf normal_mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo \ -o normal_is-report.pdf normal_is_metrics.txt # ****************************************** # 3a. Remove Duplicate Reads for tumor # sample. It is possible # to remove instead of mark duplicates # by adding the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i tumor_sorted.bam --algo LocusCollector \ --fun score_info tumor_score.txt || { echo "LocusCollector1 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i tumor_sorted.bam --algo Dedup \ --score_info tumor_score.txt --metrics tumor_dedup_metrics.txt tumor_deduped.bam || \ { echo "Dedup1 failed"; exit 1; } # ****************************************** # 3b. Remove Duplicate Reads for normal # sample. It is possible # to remove instead of mark duplicates # by adding the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i normal_sorted.bam --algo LocusCollector \ --fun score_info normal_score.txt || { echo "LocusCollector2 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i normal_sorted.bam --algo Dedup \ --score_info normal_score.txt --metrics normal_dedup_metrics.txt normal_deduped.bam || \ { echo "Dedup2 failed"; exit 1; } # ****************************************** # 4a. Somatic Variant Calling - TNhaplotyper2 # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_deduped.bam \ -i normal_deduped.bam \ --algo TNhaplotyper2 --tumor_sample $TUMOR_SM --normal_sample $NORMAL_SM \ ${PON:+--pon $PON} ${GERMLINE_VCF:+--germline_vcf $GERMLINE_VCF} output-tnhap2-tmp.vcf.gz \ --algo OrientationBias --tumor_sample $TUMOR_SM output-orientation \ --algo ContaminationModel --tumor_sample $TUMOR_SM --normal_sample $NORMAL_SM \ --vcf $CONTAMINATION_VCF \ --tumor_segments output-contamination-segments output-contamination || \ { echo "TNhaplotyper2 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA --algo TNfilter \ -v output-tnhap2-tmp.vcf.gz --tumor_sample $TUMOR_SM --normal_sample $NORMAL_SM \ --contamination output-contamination --tumor_segments output-contamination-segments \ --orientation_priors output-orientation output-tnhap2.vcf.gz || \ { echo "TNfilter failed"; exit 1; } # Uncomment the following commands to run somatic variant calling with # TNhaplotyper # ****************************************** # 4b. Somatic Variant Calling - TNhaplotyper # ****************************************** #$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_deduped.bam \ # -i normal_deduped.bam \ # --algo TNhaplotyper --tumor_sample $TUMOR_SM --normal_sample $NORMAL_SM \ # --dbsnp $KNOWN_DBSNP output-tnhaplotyper.vcf.gz || \ # { echo "TNhaplotyper failed"; exit 1; } # Uncomment the following commands to run indel realignment, corealignment and # somatic variant calling with TNsnv # ****************************************** # 5a. Indel realigner for tumor sample # ****************************************** #$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_deduped.bam \ # --algo Realigner -k $KNOWN_MILLS -k $KNOWN_INDELS tumor_realigned.bam || \ # { echo "Realigner1 failed"; exit 1; } # ****************************************** # 5b. Indel realigner for normal sample # ****************************************** #$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i normal_deduped.bam \ # --algo Realigner -k $KNOWN_MILLS -k $KNOWN_INDELS normal_realigned.bam || \ # { echo "Realigner2 failed"; exit 1; } # ****************************************** # 6. Corealignment of tumor and normal # ****************************************** #$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_realigned.bam \ # -i normal_realigned.bam \ # --algo Realigner -k $KNOWN_MILLS -k $KNOWN_INDELS tn_corealigned.bam || \ # { echo "Corealignment failed"; exit 1; } # ****************************************** # 7. Somatic Variant Calling - TNsnv # ****************************************** #$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tn_corealigned.bam \ # --algo TNsnv --tumor_sample $TUMOR_SM --normal_sample $NORMAL_SM --dbsnp $KNOWN_DBSNP \ # --call_stats_out output-call.stats output-tnsnv.vcf.gz || \ # { echo "TNsnv failed"; exit 1; } ``` ### 4.4.4 TNseq®配对样本全外显子组分析脚本 以下是 Sentieon TNseq 对肿瘤-正常样本分析的全外显子组脚本示例: ``` #!/bin/sh # Copyright (c) 2016-2024 Sentieon Inc. All rights reserved # ******************************************* # Script to perform TN seq variant calling # using a matched paired Tumor+normal sample with fastq # files named normal_1.fastq.gz, normal_2.fastq.gz # tumor_1.fastq.gz, tumor_2.fastq.gz # ******************************************* set -eu # Update with the fullpath location of your sample fastq TUMOR_SM="tumor_sample" #sample name TUMOR_RGID="rg_$TUMOR_SM" #read group ID NORMAL_SM="normal_sample" #sample name NORMAL_RGID="rg_$NORMAL_SM" #read group ID PL="ILLUMINA" #or other sequencing platform FASTQ_FOLDER="/home/pipeline/samples" TUMOR_FASTQ_1="$FASTQ_FOLDER/tumor_1.fastq.gz" TUMOR_FASTQ_2="$FASTQ_FOLDER/tumor_2.fastq.gz" #If using Illumina paired data NORMAL_FASTQ_1="$FASTQ_FOLDER/normal_1.fastq.gz" NORMAL_FASTQ_2="$FASTQ_FOLDER/normal_2.fastq.gz" # Update with the location of the reference data files FASTA_DIR="/home/regression/references/b37/" FASTA="$FASTA_DIR/human_g1k_v37_decoy.fasta" KNOWN_DBSNP="$FASTA_DIR/dbsnp_138.b37.vcf.gz" KNOWN_INDELS="$FASTA_DIR/1000G_phase1.indels.b37.vcf.gz" KNOWN_MILLS="$FASTA_DIR/Mills_and_1000G_gold_standard.indels.b37.vcf.gz" CONTAMINATION_VCF="$FASTA_DIR/germline_vcf-af-only-gnomad.raw.sites.vcf" # A VCF of germline sites to use for contamination detection PON= # the Mutect2 panel-of-normals VCF file GERMLINE_VCF= # A VCF of known germline sites INTERVAL_FILE="$FASTA_DIR/TruSeq_exome_targeted_regions.b37.bed" # Update with the location of the Sentieon software package and license file SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-|release_version| export SENTIEON_LICENSE=/home/Licenses/Sentieon.lic #or using licsrvr: c1n11.sentieon.com:5443 # Other settings NT=$(nproc) #number of threads to use in computation, set to number of cores in the server START_DIR="$PWD/test/TNseq" #Determine where the output files will be stored # You do not need to modify any of the lines below unless you want to tweak the pipeline # ************************************************************************************************************************************************************************ # ****************************************** # 0. Setup # ****************************************** WORKDIR="$START_DIR" mkdir -p $WORKDIR LOGFILE=$WORKDIR/run.log exec >$LOGFILE 2>&1 cd $WORKDIR # ****************************************** # 1a. Mapping reads with BWA-MEM, sorting for tumor sample # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$TUMOR_RGID\tSM:$TUMOR_SM\tPL:$PL" \ -t $NT -K 10000000 $FASTA $TUMOR_FASTQ_1 $TUMOR_FASTQ_2 || \ { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -o tumor_sorted.bam -t $NT --sam2bam -i - || \ { echo "Alignment1 failed"; exit 1; } # ****************************************** # 1b. Mapping reads with BWA-MEM, sorting for normal sample # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$NORMAL_RGID\tSM:$NORMAL_SM\tPL:$PL" \ -t $NT -K 10000000 $FASTA $NORMAL_FASTQ_1 $NORMAL_FASTQ_2 || \ { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -o normal_sorted.bam -t $NT --sam2bam -i - || \ { echo "Alignment2 failed"; exit 1; } # ****************************************** # 2a. Metrics for tumor sample # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT ${INTERVAL_FILE:+--interval $INTERVAL_FILE} -i tumor_sorted.bam \ --algo MeanQualityByCycle tumor_mq_metrics.txt \ --algo QualDistribution tumor_qd_metrics.txt --algo GCBias \ --summary tumor_gc_summary.txt tumor_gc_metrics.txt --algo AlignmentStat \ --adapter_seq '' tumor_aln_metrics.txt \ --algo InsertSizeMetricAlgo tumor_is_metrics.txt \ --algo CoverageMetrics --omit_base_output tumor_coverage_metrics || \ { echo "Metrics1 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o tumor_gc-report.pdf tumor_gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution \ -o tumor_qd-report.pdf tumor_qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle \ -o tumor_mq-report.pdf tumor_mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo \ -o tumor_is-report.pdf tumor_is_metrics.txt # ****************************************** # 2b. Metrics for normal sample # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT ${INTERVAL_FILE:+--interval $INTERVAL_FILE} -i normal_sorted.bam \ --algo MeanQualityByCycle normal_mq_metrics.txt \ --algo QualDistribution normal_qd_metrics.txt --algo GCBias \ --summary normal_gc_summary.txt normal_gc_metrics.txt --algo AlignmentStat \ --adapter_seq '' normal_aln_metrics.txt \ --algo InsertSizeMetricAlgo normal_is_metrics.txt \ --algo CoverageMetrics --omit_base_output normal_coverage_metrics || \ { echo "Metrics2 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o normal_gc-report.pdf normal_gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution \ -o normal_qd-report.pdf normal_qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle \ -o normal_mq-report.pdf normal_mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo \ -o normal_is-report.pdf normal_is_metrics.txt # ****************************************** # 3a. Remove Duplicate Reads for tumor # sample. It is possible # to remove instead of mark duplicates # by adding the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i tumor_sorted.bam --algo LocusCollector \ --fun score_info tumor_score.txt || { echo "LocusCollector1 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i tumor_sorted.bam --algo Dedup \ --score_info tumor_score.txt --metrics tumor_dedup_metrics.txt tumor_deduped.bam || \ { echo "Dedup1 failed"; exit 1; } # ****************************************** # 3b. Remove Duplicate Reads for normal # sample. It is possible # to remove instead of mark duplicates # by adding the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i normal_sorted.bam --algo LocusCollector \ --fun score_info normal_score.txt || { echo "LocusCollector2 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i normal_sorted.bam --algo Dedup \ --score_info normal_score.txt --metrics normal_dedup_metrics.txt normal_deduped.bam || \ { echo "Dedup2 failed"; exit 1; } # ****************************************** # 4a. Somatic Variant Calling - TNhaplotyper2 # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT ${INTERVAL_FILE:+--interval $INTERVAL_FILE} -i tumor_deduped.bam \ -i normal_deduped.bam \ --algo TNhaplotyper2 --tumor_sample $TUMOR_SM --normal_sample $NORMAL_SM \ ${PON:+--pon $PON} ${GERMLINE_VCF:+--germline_vcf $GERMLINE_VCF} output-tnhap2-tmp.vcf.gz \ --algo OrientationBias --tumor_sample $TUMOR_SM output-orientation \ --algo ContaminationModel --tumor_sample $TUMOR_SM --normal_sample $NORMAL_SM \ --vcf $CONTAMINATION_VCF \ --tumor_segments output-contamination-segments output-contamination || \ { echo "TNhaplotyper2 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA --algo TNfilter \ -v output-tnhap2-tmp.vcf.gz --tumor_sample $TUMOR_SM --normal_sample $NORMAL_SM \ --contamination output-contamination --tumor_segments output-contamination-segments \ --orientation_priors output-orientation output-tnhap2.vcf.gz || \ { echo "TNfilter failed"; exit 1; } # Uncomment the following commands to run somatic variant calling with # TNhaplotyper # ****************************************** # 4b. Somatic Variant Calling - TNhaplotyper # ****************************************** #$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT ${INTERVAL_FILE:+--interval $INTERVAL_FILE} -i tumor_deduped.bam \ # -i normal_deduped.bam \ # --algo TNhaplotyper --tumor_sample $TUMOR_SM --normal_sample $NORMAL_SM \ # --dbsnp $KNOWN_DBSNP output-tnhaplotyper.vcf.gz || \ # { echo "TNhaplotyper failed"; exit 1; } # Uncomment the following commands to run indel realignment, corealignment and # somatic variant calling with TNsnv # ****************************************** # 5a. Indel realigner for tumor sample # ****************************************** #$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_deduped.bam \ # --algo Realigner -k $KNOWN_MILLS -k $KNOWN_INDELS tumor_realigned.bam || \ # { echo "Realigner1 failed"; exit 1; } # ****************************************** # 5b. Indel realigner for normal sample # ****************************************** #$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i normal_deduped.bam \ # --algo Realigner -k $KNOWN_MILLS -k $KNOWN_INDELS normal_realigned.bam || \ # { echo "Realigner2 failed"; exit 1; } # ****************************************** # 6. Corealignment of tumor and normal # ****************************************** #$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_realigned.bam \ # -i normal_realigned.bam \ # --algo Realigner -k $KNOWN_MILLS -k $KNOWN_INDELS tn_corealigned.bam || \ # { echo "Corealignment failed"; exit 1; } # ****************************************** # 7. Somatic Variant Calling - TNsnv # ****************************************** #$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT ${INTERVAL_FILE:+--interval $INTERVAL_FILE} -i tn_corealigned.bam \ # --algo TNsnv --tumor_sample $TUMOR_SM --normal_sample $NORMAL_SM --dbsnp $KNOWN_DBSNP \ # --call_stats_out output-call.stats output-tnsnv.vcf.gz || \ # { echo "TNsnv failed"; exit 1; } ``` ### 4.4.5 TNseq®单样本分析脚本 以下是Sentieon TNseq在只使用肿瘤样本,没有配对正常样本时的全基因组分析脚本示例: ``` #!/bin/sh # Copyright (c) 2016-2024 Sentieon Inc. All rights reserved # ******************************************* # Script to perform TN seq variant calling # using a Tumor sample with fastq files # named tumor_1.fastq.gz, tumor_2.fastq.gz # and data from a Panel of Normals and Cosmic DB # ******************************************* set -eu # Update with the fullpath location of your sample fastq TUMOR_SM="tumor_sample" #sample name TUMOR_RGID="rg_$TUMOR_SM" #read group ID PL="ILLUMINA" #or other sequencing platform FASTQ_FOLDER="/home/pipeline/samples" TUMOR_FASTQ_1="$FASTQ_FOLDER/tumor_1.fastq.gz" TUMOR_FASTQ_2="$FASTQ_FOLDER/tumor_2.fastq.gz" #If using Illumina paired data # Update with the location of the reference data files FASTA_DIR="/home/regression/references/b37/" FASTA="$FASTA_DIR/human_g1k_v37_decoy.fasta" KNOWN_DBSNP="$FASTA_DIR/dbsnp_138.b37.vcf.gz" KNOWN_INDELS="$FASTA_DIR/1000G_phase1.indels.b37.vcf.gz" KNOWN_MILLS="$FASTA_DIR/Mills_and_1000G_gold_standard.indels.b37.vcf.gz" # Update with the location of the panel of normal and CosmicDB vcf files # We recommend that you create the panel of normal file with the corresponding algorithm that you plan to use for the somatic mutation calling. PANEL_OF_NORMAL_TNSNV= PANEL_OF_NORMAL_TNHAPLOTYPER= PANEL_OF_NORMAL_TNHAPLOTYPER2= COSMIC_DB="/home/regression/references/b37/b37_cosmic_v54_120711.vcf.gz" CONTAMINATION_VCF="$FASTA_DIR/germline_vcf-af-only-gnomad.raw.sites.vcf" # A VCF of germline sites to use for contamination detection GERMLINE_VCF= # A VCF of known germline sites # Update with the location of the Sentieon software package and license file SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-|release_version| export SENTIEON_LICENSE=/home/Licenses/Sentieon.lic #or using licsrvr: c1n11.sentieon.com:5443 # Other settings NT=$(nproc) #number of threads to use in computation, set to number of cores in the server START_DIR="$PWD/test/TNseq_tumoronly" #Determine where the output files will be stored # You do not need to modify any of the lines below unless you want to tweak the pipeline # ************************************************************************************************************************************************************************ # ****************************************** # 0. Setup # ****************************************** WORKDIR="$START_DIR" mkdir -p $WORKDIR LOGFILE=$WORKDIR/run.log exec >$LOGFILE 2>&1 cd $WORKDIR # ****************************************** # 1a. Mapping reads with BWA-MEM, sorting for tumor sample # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$TUMOR_RGID\tSM:$TUMOR_SM\tPL:$PL" \ -t $NT -K 10000000 $FASTA $TUMOR_FASTQ_1 $TUMOR_FASTQ_2 || \ { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -o tumor_sorted.bam -t $NT --sam2bam -i - || \ { echo "Alignment failed"; exit 1; } # ****************************************** # 2a. Metrics for tumor sample # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_sorted.bam \ --algo MeanQualityByCycle tumor_mq_metrics.txt \ --algo QualDistribution tumor_qd_metrics.txt --algo GCBias \ --summary tumor_gc_summary.txt tumor_gc_metrics.txt --algo AlignmentStat \ --adapter_seq '' tumor_aln_metrics.txt \ --algo InsertSizeMetricAlgo tumor_is_metrics.txt \ --algo CoverageMetrics --omit_base_output tumor_coverage_metrics || \ { echo "Metrics failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o tumor_gc-report.pdf tumor_gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution \ -o tumor_qd-report.pdf tumor_qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle \ -o tumor_mq-report.pdf tumor_mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo \ -o tumor_is-report.pdf tumor_is_metrics.txt # ****************************************** # 3a. Remove Duplicate Reads for tumor # sample. It is possible # to mark instead of remove duplicates # by ommiting the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i tumor_sorted.bam --algo LocusCollector \ --fun score_info tumor_score.txt || { echo "LocusCollector failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i tumor_sorted.bam --algo Dedup \ --rmdup --score_info tumor_score.txt --metrics tumor_dedup_metrics.txt tumor_deduped.bam || \ { echo "Dedup failed"; exit 1; } # ****************************************** # 4a. Somatic Variant Calling - TNhaplotyper2 # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_deduped.bam \ --algo TNhaplotyper2 --tumor_sample $TUMOR_SM \ ${PANEL_OF_NORMAL_TNHAPLOTYPER2:+--pon $PANEL_OF_NORMAL_TNHAPLOTYPER2} \ ${GERMLINE_VCF:+--germline_vcf $GERMLINE_VCF} output-tnhap2-tmp.vcf.gz \ --algo OrientationBias --tumor_sample $TUMOR_SM output-orientation \ --algo ContaminationModel --tumor_sample $TUMOR_SM --vcf $CONTAMINATION_VCF \ --tumor_segments output-contamination-segments output-contamination || \ { echo "TNhaplotyper2 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA --algo TNfilter \ -v output-tnhap2-tmp.vcf.gz --tumor_sample $TUMOR_SM \ --contamination output-contamination --tumor_segments output-contamination-segments \ --orientation_priors output-orientation output-tnhap2.vcf.gz || \ { echo "TNfilter failed"; exit 1; } # Uncomment the following commands to run somatic variant calling with TNhaplotyper # ****************************************** # 4b. Somatic Variant Calling - TNhaplotyper # ****************************************** #$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_deduped.bam \ # --algo TNhaplotyper --tumor_sample $TUMOR_SM \ # ${PANEL_OF_NORMAL_TNHAPLOTYPER:+--pon $PANEL_OF_NORMAL_TNHAPLOTYPER} \ # --cosmic $COSMIC_DB --dbsnp $KNOWN_DBSNP \ # output-tnhaplotyper.vcf.gz || { echo "TNhaplotyper failed"; exit 1; } # Uncomment the following commands to run indel realignment, and somatic variant # calling with TNsnv # ****************************************** # 5. Indel realigner for tumor sample # ****************************************** #$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_deduped.bam \ # --algo Realigner -k $KNOWN_MILLS -k $KNOWN_INDELS tumor_realigned.bam || \ # { echo "Realigner failed"; exit 1; } # ****************************************** # 6. Somatic Variant Calling - TNsnv # ****************************************** #$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_realigned.bam \ # --algo TNsnv --tumor_sample $TUMOR_SM \ # ${PANEL_OF_NORMAL_TNSNV:+--pon $PANEL_OF_NORMAL_TNSNV} \ # --cosmic $COSMIC_DB --dbsnp $KNOWN_DBSNP \ # --call_stats_out output-call.stats output-tnsnv.vcf.gz || \ # { echo "TNsnv failed"; exit 1; } ``` ### 4.4.6 TNseq® 单样本全外显子组分析脚本 以下是 Sentieon TNseq 在只使用肿瘤样本,没有配对正常样本时的全外显子组分析脚本示例: ``` #!/bin/sh # Copyright (c) 2016-2024 Sentieon Inc. All rights reserved # ******************************************* # Script to perform TN seq variant calling # using a Tumor sample with fastq files # named tumor_1.fastq.gz, tumor_2.fastq.gz # and data from a Panel of Normals and Cosmic DB # ******************************************* set -eu # Update with the fullpath location of your sample fastq TUMOR_SM="tumor_sample" #sample name TUMOR_RGID="rg_$TUMOR_SM" #read group ID PL="ILLUMINA" #or other sequencing platform FASTQ_FOLDER="/home/pipeline/samples" TUMOR_FASTQ_1="$FASTQ_FOLDER/tumor_1.fastq.gz" TUMOR_FASTQ_2="$FASTQ_FOLDER/tumor_2.fastq.gz" #If using Illumina paired data # Update with the location of the reference data files FASTA_DIR="/home/regression/references/b37/" FASTA="$FASTA_DIR/human_g1k_v37_decoy.fasta" KNOWN_DBSNP="$FASTA_DIR/dbsnp_138.b37.vcf.gz" KNOWN_INDELS="$FASTA_DIR/1000G_phase1.indels.b37.vcf.gz" KNOWN_MILLS="$FASTA_DIR/Mills_and_1000G_gold_standard.indels.b37.vcf.gz" # Update with the location of the panel of normal and CosmicDB vcf files # We recommend that you create the panel of normal file with the corresponding algorithm that you plan to use for the somatic mutation calling. PANEL_OF_NORMAL_TNSNV= PANEL_OF_NORMAL_TNHAPLOTYPER= PANEL_OF_NORMAL_TNHAPLOTYPER2= COSMIC_DB="/home/regression/references/b37/b37_cosmic_v54_120711.vcf.gz" CONTAMINATION_VCF="$FASTA_DIR/germline_vcf-af-only-gnomad.raw.sites.vcf" # A VCF of germline sites to use for contamination detection GERMLINE_VCF= # A VCF of known germline sites INTERVAL_FILE="$FASTA_DIR/TruSeq_exome_targeted_regions.b37.bed" # Update with the location of the Sentieon software package and license file SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-|release_version| export SENTIEON_LICENSE=/home/Licenses/Sentieon.lic #or using licsrvr: c1n11.sentieon.com:5443 # Other settings NT=$(nproc) #number of threads to use in computation, set to number of cores in the server START_DIR="$PWD/test/TNseq_tumoronly" #Determine where the output files will be stored # You do not need to modify any of the lines below unless you want to tweak the pipeline # ************************************************************************************************************************************************************************ # ****************************************** # 0. Setup # ****************************************** WORKDIR="$START_DIR" mkdir -p $WORKDIR LOGFILE=$WORKDIR/run.log exec >$LOGFILE 2>&1 cd $WORKDIR # ****************************************** # 1a. Mapping reads with BWA-MEM, sorting for tumor sample # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$TUMOR_RGID\tSM:$TUMOR_SM\tPL:$PL" \ -t $NT -K 10000000 $FASTA $TUMOR_FASTQ_1 $TUMOR_FASTQ_2 || \ { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -o tumor_sorted.bam -t $NT --sam2bam -i - || \ { echo "Alignment failed"; exit 1; } # ****************************************** # 2a. Metrics for tumor sample # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT ${INTERVAL_FILE:+--interval $INTERVAL_FILE} -i tumor_sorted.bam \ --algo MeanQualityByCycle tumor_mq_metrics.txt \ --algo QualDistribution tumor_qd_metrics.txt --algo GCBias \ --summary tumor_gc_summary.txt tumor_gc_metrics.txt --algo AlignmentStat \ --adapter_seq '' tumor_aln_metrics.txt \ --algo InsertSizeMetricAlgo tumor_is_metrics.txt \ --algo CoverageMetrics --omit_base_output tumor_coverage_metrics || \ { echo "Metrics failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o tumor_gc-report.pdf tumor_gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution \ -o tumor_qd-report.pdf tumor_qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle \ -o tumor_mq-report.pdf tumor_mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo \ -o tumor_is-report.pdf tumor_is_metrics.txt # ****************************************** # 3a. Remove Duplicate Reads for tumor # sample. It is possible # to mark instead of remove duplicates # by ommiting the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i tumor_sorted.bam --algo LocusCollector \ --fun score_info tumor_score.txt || { echo "LocusCollector failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i tumor_sorted.bam --algo Dedup \ --rmdup --score_info tumor_score.txt --metrics tumor_dedup_metrics.txt tumor_deduped.bam || \ { echo "Dedup failed"; exit 1; } # ****************************************** # 4a. Somatic Variant Calling - TNhaplotyper2 # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT ${INTERVAL_FILE:+--interval $INTERVAL_FILE} -i tumor_deduped.bam \ --algo TNhaplotyper2 --tumor_sample $TUMOR_SM \ ${PANEL_OF_NORMAL_TNHAPLOTYPER2:+--pon $PANEL_OF_NORMAL_TNHAPLOTYPER2} \ ${GERMLINE_VCF:+--germline_vcf $GERMLINE_VCF} output-tnhap2-tmp.vcf.gz \ --algo OrientationBias --tumor_sample $TUMOR_SM output-orientation \ --algo ContaminationModel --tumor_sample $TUMOR_SM --vcf $CONTAMINATION_VCF \ --tumor_segments output-contamination-segments output-contamination || \ { echo "TNhaplotyper2 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA --algo TNfilter \ -v output-tnhap2-tmp.vcf.gz --tumor_sample $TUMOR_SM \ --contamination output-contamination --tumor_segments output-contamination-segments \ --orientation_priors output-orientation output-tnhap2.vcf.gz || \ { echo "TNfilter failed"; exit 1; } # Uncomment the following commands to run somatic variant calling with TNhaplotyper # ****************************************** # 4b. Somatic Variant Calling - TNhaplotyper # ****************************************** #$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT ${INTERVAL_FILE:+--interval $INTERVAL_FILE} -i tumor_deduped.bam \ # --algo TNhaplotyper --tumor_sample $TUMOR_SM \ # ${PANEL_OF_NORMAL_TNHAPLOTYPER:+--pon $PANEL_OF_NORMAL_TNHAPLOTYPER} \ # --cosmic $COSMIC_DB --dbsnp $KNOWN_DBSNP \ # output-tnhaplotyper.vcf.gz || { echo "TNhaplotyper failed"; exit 1; } # Uncomment the following commands to run indel realignment, and somatic variant # calling with TNsnv # ****************************************** # 5. Indel realigner for tumor sample # ****************************************** #$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_deduped.bam \ # --algo Realigner -k $KNOWN_MILLS -k $KNOWN_INDELS tumor_realigned.bam || \ # { echo "Realigner failed"; exit 1; } # ****************************************** # 6. Somatic Variant Calling - TNsnv # ****************************************** #$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT ${INTERVAL_FILE:+--interval $INTERVAL_FILE} -i tumor_realigned.bam \ # --algo TNsnv --tumor_sample $TUMOR_SM \ # ${PANEL_OF_NORMAL_TNSNV:+--pon $PANEL_OF_NORMAL_TNSNV} \ # --cosmic $COSMIC_DB --dbsnp $KNOWN_DBSNP \ # --call_stats_out output-call.stats output-tnsnv.vcf.gz || \ # { echo "TNsnv failed"; exit 1; } ``` ## 4.5 TNscope® Sentieon® Genomics 软件的用途是执行生信分析流程,用于仅使用肿瘤或肿瘤-正常对照分析的体细胞变异和结构变异检测算法。图4-3 说明了这样一个典型的生信分析流程。 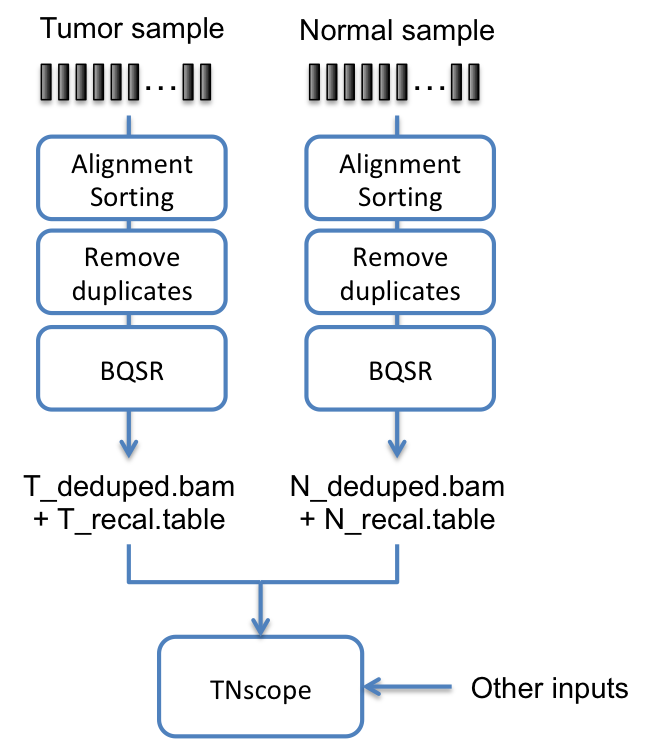 <center>图4-3 基于 TNscope® 的肿瘤-正常对照分析的推荐流程</center> <br> 请查看 https://support.sentieon.com/appnotes/somatic 中的应用说明以获取推荐的具有不同输入的体细胞变异检测流程。 ### 4.5.1 概述 在此生物信息学流程中,您将需要以下输入: - FASTA 文件:包含与待分析样本对应的参考基因组核苷酸序列。 - 两组 FASTQ 文件:包含待分析样本的核苷酸序列,一组用于肿瘤样本,一组用于匹配的正常样本。这些文件包含了 DNA 测序的原始读段。软件支持输入经过 GZIP 压缩的 FASTQ 文件。软件仅支持包含 Sanger 格式(Phred+33)质量分数的文件。 - (可选)dbSNP 数据库:您希望包含在流程中的单核苷酸多态性数据库数据,以 VCF 文件形式使用。 - (可选)已知位点集合:您希望包含在流程中的多个已知位点集合,以 VCF 文件形式使用。 肿瘤-正常匹配对的生信分析流程包括以下步骤: 1. 使用 DNA 独立预处理肿瘤和正常样品 seq 管道(如 第 4.1.2 节 中介绍的流程)具有以下阶段: - 将读段映射到参考基因组;您需要确保肿瘤样本和正常样本的 SM 样本标签不同,因为在体细胞变异检测中需要将其作为参数。同时,您还需要确保两个样本的 RG:ID是不同且唯一的。 - 计算数据指标。 - 移除重复序列。 - (可选)碱基质量分数重校准(BQSR)。 2. 体细胞变异检测:此步骤识别癌症基因组数据相对于正常基因组表现出体细胞变异的位点,并计算该位点的基因型。 ### 4.5.2 TNscope® 使用步骤 对于映射、重复去除和碱基质量分数重校准阶段,请参考 *第4.1.2 节* 获取详细的使用说明。 #### (1)有或没有匹配正常样本的体细胞变异发现 运行单条命令以对肿瘤-正常配对对照样本进行变异检测: ``` sentieon driver -t NUMBER_THREADS -r REFERENCE \ -i TUMOR_DEDUPED_BAM [-q TUMOR_RECAL_DATA.TABLE] \ -i NORMAL_DEDUPED_BAM [-q NORMAL_RECAL_DATA.TABLE] \ --algo TNscope \ --tumor_sample TUMOR_SAMPLE_NAME --normal_sample NORMAL_SAMPLE_NAME \ [--dbsnp DBSNP] OUT_TN_VCF ``` 如果您没有匹配的正常样本,则可以跳过正常样本 BAM 文件及样本名称输入,这些参数不是必需的;但请注意,由于缺乏正常样本对照,种系变异将存在于输出文件中: ``` sentieon driver -t NUMBER_THREADS -r REFERENCE \ -i TUMOR_DEDUPED_BAM [-q TUMOR_RECAL_DATA.TABLE] \ --algo TNscope --tumor_sample TUMOR_SAMPLE_NAME \ [--dbsnp DBSNP] OUT_TN_VCF ``` 为了在缺少匹配的正常样本时,您可以将匹配的正常样本替换为使用下文*第4.5.2节(2)生成正常 VCF 面板文件* 中描述的方法生成的通用正常面板。 运行单条命令以仅对肿瘤样本进行变异检测: ``` sentieon driver -t NUMBER_THREADS -r REFERENCE \ -i TUMOR_DEDUPED_BAM [-q TUMOR_RECAL_DATA.TABLE] \ --algo TNscope --tumor_sample TUMOR_SAMPLE_NAME \ --pon PANEL_OF_NORMAL_VCF [--dbsnp DBSNP] OUT_TN_VCF ``` 该命令需要以下输入参数: - `NUMBER_THREADS`:计算中将使用的线程数。建议该数量不要超过系统中可用的计算核心数。 - `REFERENCE`:参考FASTA文件的路径。需确保参考序列与映射阶段使用的完全一致。 - `TUMOR_DEDUPED_BAM`:肿瘤样本经过预处理(去重后)的 BAM 文件路径。 - `NORMAL_DEDUPED_BAM`:正常样本经过预处理(去重后)的 BAM 文件路径。 - `TUMOR_SAMPLE_NAME`:在“映射读段至参考基因组”阶段为肿瘤样本设置的样本名称。 - `OUT_TN_VCF`:包含变异检测的输出文件的路径和文件名。 该命令的以下输入参数是可选的: - `TUMOR_RECAL_DATA.TABLE`:肿瘤样本 BQSR 阶段存储结果的路径。 - `NORMAL_RECAL_DATA.TABLE`:正常样本 BQSR 阶段存储结果的路径。 - `NORMAL_SAMPLE_NAME`:在“映射读段至参考基因组”阶段为正常样本设置的样本名称。 - `DBSNP`:单核苷酸多态性数据库(dbSNP)的位置。dbSNP 中的变异更有可能被标记为胚系,因为它们需要更多证据证明是体细胞变异。您只能使用一个 dbSNP 文件。 - `PANEL_OF_NORMAL_VCF`:正常样本面板 VCF 文件的路径和名称。 #### (2)生成正常 VCF 面板文件 为了生成适用于 TNscope® 的自有正常样本对照集 VCF 文件,您需要对面板中的每个正常样本运行以下命令: ``` sentieon driver -t NUMBER_THREADS -r REFERENCE -i NORMAL_DEDUPED_BAM \ [-q NORMAL_RECAL_DATA.TABLE] --algo TNscope \ --tumor_sample NORMAL_SAMPLE_NAME OUT_NORMAL_VCF ``` 该命令需要以下输入参数: - `NUMBER_THREADS`:计算中将使用的线程数。建议该数量不要超过系统中可用的计算核心数。 - `REFERENCE`:参考FASTA文件的路径。需确保参考序列与映射阶段使用的完全一致。 - `NORMAL_DEDUPED_BAM`:正常样本经过预处理(去重后)的 BAM 文件路径。 - `NORMAL_SAMPLE_NAME`:在映射阶段为该正常样本设置的样本名称。 - `OUT_NORMAL_VCF`:包含该输入正常样本相关变异的输出 VCF 文件路径及文件名。 该命令的以下输入数据是可选的: - `NORMAL_RECAL_DATA.TABLE`:正常样本 BQSR 阶段存储结果的路径。 生成包含在面板中的所有 VCF 文件后,您需要将它们合并到一个正常 VCF 面板中。您可以使用 bcftools 实现该目的: ``` BCF=/path_to_bcftools export BCFTOOLS_PLUGINS=$BCF/plugins DIR=/path_to_normal_vcf_file $BCF/bcftools merge -m all -f PASS,. --force-samples $DIR/*.vcf.gz |\ $BCF/bcftools plugin fill-AN-AC |\ $BCF/bcftools filter -i 'SUM(AC)>1' > panel_of_normal.vcf ``` ### 4.5.3 TNscope® PCR扩增子分析脚本 以下是TNscope使用配对正常样本对扩增子数据进行体细胞变异检测的脚本示例: ``` #!/bin/sh # Copyright (c) 2016-2024 Sentieon Inc. All rights reserved # ******************************************* # Script to perform TNscope variant calling on # PCR amplicon tumor sample with coverage over # 200x and a matched normal sample. # ******************************************* set -eu # Update with the fullpath location of your sample fastq TUMOR_SM="tumor_sample" #sample name TUMOR_RGID="rg_$TUMOR_SM" #read group ID NORMAL_SM="normal_sample" #sample name NORMAL_RGID="rg_$NORMAL_SM" #read group ID PL="ILLUMINA" #or other sequencing platform FASTQ_FOLDER="/home/pipeline/samples" TUMOR_FASTQ_1="$FASTQ_FOLDER/tumor_1.fastq.gz" TUMOR_FASTQ_2="$FASTQ_FOLDER/tumor_2.fastq.gz" #If using Illumina paired data NORMAL_FASTQ_1="$FASTQ_FOLDER/normal_1.fastq.gz" NORMAL_FASTQ_2="$FASTQ_FOLDER/normal_2.fastq.gz" #If using Illumina paired data # Update with the location of the reference data files FASTA_DIR="/home/regression/references/b37" FASTA="$FASTA_DIR/human_g1k_v37_decoy.fasta" KNOWN_DBSNP="$FASTA_DIR/dbsnp_138.b37.vcf.gz" INTERVAL_FILE="targeted_regions.bed" # Update with the location of the Sentieon software package and license file SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-|release_version| TNSCOPE_FILTER=/home/sentieon-scripts/tnscope_filter/tnscope_filter.py export SENTIEON_LICENSE=/home/Licenses/Sentieon.lic #or using licsrvr: c1n11.sentieon.com:5443 # Other settings NT=$(nproc) #number of threads to use in computation, set to number of cores in the server START_DIR="$PWD/test/TNscope" #Determine where the output files will be stored # ************************************************************************************************************************************************************************ # ****************************************** # 0. Setup # ****************************************** WORKDIR="$START_DIR" mkdir -p $WORKDIR LOGFILE=$WORKDIR/run.log exec >$LOGFILE 2>&1 cd $WORKDIR # ****************************************** # 1a. Mapping reads with BWA-MEM, sorting for the tumor sample # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$TUMOR_RGID\tSM:$TUMOR_SM\tPL:$PL" \ -t $NT -K 10000000 $FASTA $TUMOR_FASTQ_1 $TUMOR_FASTQ_2 || \ { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -o tumor_sorted.bam -t $NT --sam2bam -i - || \ { echo "Alignment1 failed"; exit 1; } # ****************************************** # 1b. Mapping reads with BWA-MEM, sorting for the normal sample # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$NORMAL_RGID\tSM:$NORMAL_SM\tPL:$PL" \ -t $NT -K 10000000 $FASTA $NORMAL_FASTQ_1 $NORMAL_FASTQ_2 || \ { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -o normal_sorted.bam -t $NT --sam2bam -i - || \ { echo "Alignment2 failed"; exit 1; } # ****************************************** # 2a. Metrics for the tumor sample # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_sorted.bam \ --algo MeanQualityByCycle tumor_mq_metrics.txt \ --algo QualDistribution tumor_qd_metrics.txt --algo GCBias \ --summary tumor_gc_summary.txt tumor_gc_metrics.txt --algo AlignmentStat \ --adapter_seq '' tumor_aln_metrics.txt \ --algo InsertSizeMetricAlgo tumor_is_metrics.txt \ --algo CoverageMetrics --omit_base_output tumor_coverage_metrics || \ { echo "Metrics1 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o tumor_gc-report.pdf tumor_gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution \ -o tumor_qd-report.pdf tumor_qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle \ -o tumor_mq-report.pdf tumor_mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo \ -o tumor_is-report.pdf tumor_is_metrics.txt # ****************************************** # 2b. Metrics for the normal sample # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i normal_sorted.bam \ --algo MeanQualityByCycle normal_mq_metrics.txt \ --algo QualDistribution normal_qd_metrics.txt --algo GCBias \ --summary normal_gc_summary.txt normal_gc_metrics.txt --algo AlignmentStat \ --adapter_seq '' normal_aln_metrics.txt \ --algo InsertSizeMetricAlgo normal_is_metrics.txt \ --algo CoverageMetrics --omit_base_output normal_coverage_metrics || \ { echo "Metrics2 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o normal_gc-report.pdf normal_gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution \ -o normal_qd-report.pdf normal_qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle \ -o normal_mq-report.pdf normal_mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo \ -o normal_is-report.pdf normal_is_metrics.txt # ****************************************** # 4. Somatic and Structural variant calling # ****************************************** # Consider adding `--disable_detector sv` if not interested in SV calling $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT \ ${INTERVAL_FILE:+--interval_padding 10 --interval $INTERVAL_FILE} \ -i tumor_sorted.bam -i normal_sorted.bam \ --algo TNscope \ --tumor_sample $TUMOR_SM --normal_sample $NORMAL_SM \ --dbsnp $KNOWN_DBSNP \ --min_tumor_allele_frac 0.009 \ --max_fisher_pv_active 0.05 \ --max_normal_alt_cnt 10 \ --max_normal_alt_frac 0.01 \ --max_normal_alt_qsum 250 \ --sv_mask_ext 10 \ --prune_factor 0 \ --assemble_mode 2 \ output_tnscope.pre_filter.vcf.gz || \ { echo "TNscope failed"; exit 1; } # ****************************************** # 6. Variant filtration # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon pyexec $TNSCOPE_FILTER \ -v output_tnscope.pre_filter.vcf.gz \ --tumor_sample $TUMOR_SM \ -x amplicon --min_tumor_af 0.0095 --min_depth 200 \ output_tnscope.filter.vcf.gz || \ { echo "TNscope filter failed"; exit 1; } ``` ### 4.5.4 TNscope® UMI标签分析脚本 以下是TNscope在专门处理带有UMI标签的样本数据的脚本示例: ``` #!/bin/sh # Copyright (c) 2016-2024 Sentieon Inc. All rights reserved # ******************************************* # Script to perform TNscope variant calling on # UMI-tagged samples # ******************************************* set -eu # Update with the fullpath location of your sample fastq TUMOR_SM="tumor_sample" #sample name TUMOR_RGID="rg_$TUMOR_SM" #read group ID PL="ILLUMINA" #or other sequencing platform FASTQ_FOLDER="/home/pipeline/samples" TUMOR_FASTQ_1="$FASTQ_FOLDER/tumor_1.fastq.gz" TUMOR_FASTQ_2="$FASTQ_FOLDER/tumor_2.fastq.gz" #If using Illumina paired data # Update with the location of the reference data files FASTA_DIR="/home/regression/references/b37" FASTA="$FASTA_DIR/human_g1k_v37_decoy.fasta" KNOWN_DBSNP="$FASTA_DIR/dbsnp_138.b37.vcf.gz" INTERVAL_FILE="targeted_regions.bed" # Update with the location of the Sentieon software package and license file SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-|release_version| TNSCOPE_FILTER=/home/sentieon-scripts/tnscope_filter/tnscope_filter.py export SENTIEON_LICENSE=/home/Licenses/Sentieon.lic #or using licsrvr: c1n11.sentieon.com:5443 #UMI information READ_STRUCTURE="12M11S+T,+T" #an example duplex: "3M2S+T,3M2S+T" where duplex UMI extraction requires an identical read structure for both strands DUPLEX_UMI="false" #set to "true" if duplex # Other settings NT=$(nproc) #number of threads to use in computation, set to number of cores in the server START_DIR="$PWD/test/TNscope_umi" #Determine where the output files will be stored # ****************************************** # 0. Setup # ****************************************** WORKDIR="$START_DIR" mkdir -p $WORKDIR LOGFILE=$WORKDIR/run.log exec >$LOGFILE 2>&1 cd $WORKDIR # ****************************************** # 1. Pre-processing of FASTQ containing UMIs # ****************************************** if [ "$DUPLEX_UMI" = "true" ] ; then READ_STRUCTURE="-d $READ_STRUCTURE" fi ( $SENTIEON_INSTALL_DIR/bin/sentieon umi extract $READ_STRUCTURE $TUMOR_FASTQ_1 $TUMOR_FASTQ_2 || \ { echo -n 'Extract error' >&2; exit -1; } ) | \ ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -p -C \ -R "@RG\tID:$TUMOR_RGID\tSM:$TUMOR_SM\tPL:$PL" -t $NT \ -K 10000000 $FASTA - || { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -t $NT -r $FASTA --sam2bam -i - \ -o tumor_sorted.bam || \ { echo "Alignment failed"; exit 1; } # ****************************************** # 2. Remove duplicate reads for the tumor sample # use the consensus-based approach with UMI. # It is possible to remove instead of mark # duplicates by adding the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i tumor_sorted.bam --algo LocusCollector \ --consensus --umi_tag XR --fun score_info tumor_score.txt || { echo "LocusCollector failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i tumor_sorted.bam -r $FASTA --algo Dedup \ --score_info tumor_score.txt --metrics umi.dedup_metrics.txt tumor_deduped.bam || \ { echo "Dedup failed"; exit 1; } # ****************************************** # 3. Somatic and Structural variant calling # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_deduped.bam \ ${INTERVAL_FILE:+--interval $INTERVAL_FILE} \ --algo TNscope \ --tumor_sample $TUMOR_SM \ --dbsnp $KNOWN_DBSNP \ --disable_detector sv \ --min_tumor_allele_frac 3e-3 \ --min_tumor_lod 3.0 \ --min_init_tumor_lod 1.0 \ --assemble_mode 4 \ --pcr_indel_model NONE \ --min_base_qual 40 \ --resample_depth 100000 \ output_tnscope.pre_filter.vcf.gz || \ { echo "TNscope failed"; exit 1; } # ****************************************** # 4. Variant filtration # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon pyexec $TNSCOPE_FILTER \ -v output_tnscope.pre_filter.vcf.gz \ --tumor_sample $TUMOR_SM \ -x ctdna --min_tumor_af 0.001 --min_depth 1000 \ output_tnscope.filter.vcf.gz || \ { echo "TNscope filter failed"; exit 1; } ``` ### 4.5.5 TNscope® cfDNA分析脚本 以下是TNscope在无UMI游离DNA样本的脚本示例: ``` #!/bin/sh # Copyright (c) 2016-2024 Sentieon Inc. All rights reserved # ******************************************* # Script to perform TNscope variant calling on # hybridization captured cfDNA tumor sample # without UMI. # ******************************************* set -eu # Update with the fullpath location of your sample fastq TUMOR_SM="tumor_sample" #sample name TUMOR_RGID="rg_$TUMOR_SM" #read group ID PL="ILLUMINA" #or other sequencing platform FASTQ_FOLDER="/home/pipeline/samples" TUMOR_FASTQ_1="$FASTQ_FOLDER/tumor_1.fastq.gz" TUMOR_FASTQ_2="$FASTQ_FOLDER/tumor_2.fastq.gz" #If using Illumina paired data # Update with the location of the reference data files FASTA_DIR="/home/regression/references/b37" FASTA="$FASTA_DIR/human_g1k_v37_decoy.fasta" KNOWN_DBSNP="$FASTA_DIR/dbsnp_138.b37.vcf.gz" INTERVAL_FILE="targeted_regions.bed" # Update with the location of the Sentieon software package and license file SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-|release_version| TNSCOPE_FILTER=/home/sentieon-scripts/tnscope_filter/tnscope_filter.py export SENTIEON_LICENSE=/home/Licenses/Sentieon.lic #or using licsrvr: c1n11.sentieon.com:5443 # Other settings NT=$(nproc) #number of threads to use in computation, set to number of cores in the server START_DIR="$PWD/test/TNscope" #Determine where the output files will be stored # ************************************************************************************************************************************************************************ # ****************************************** # 0. Setup # ****************************************** WORKDIR="$START_DIR" mkdir -p $WORKDIR LOGFILE=$WORKDIR/run.log exec >$LOGFILE 2>&1 cd $WORKDIR # ****************************************** # 1. Mapping reads with BWA-MEM, sorting for the tumor sample # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$TUMOR_RGID\tSM:$TUMOR_SM\tPL:$PL" \ -t $NT -K 10000000 $FASTA $TUMOR_FASTQ_1 $TUMOR_FASTQ_2 || \ { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -o tumor_sorted.bam -t $NT --sam2bam -i - || \ { echo "Alignment failed"; exit 1; } # ****************************************** # 2. Remove duplicate reads for the tumor sample # use the consensus-based approach. # It is possible to remove instead of mark # duplicates by adding the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i tumor_sorted.bam --algo LocusCollector \ --consensus --fun score_info tumor_score.txt || { echo "LocusCollector failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i tumor_sorted.bam -r $FASTA --algo Dedup \ --score_info tumor_score.txt tumor_deduped.bam || \ { echo "Dedup failed"; exit 1; } # ****************************************** # 3. Somatic and Structural variant calling # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_deduped.bam \ ${INTERVAL_FILE:+--interval $INTERVAL_FILE} \ --algo TNscope \ --tumor_sample $TUMOR_SM \ --dbsnp $KNOWN_DBSNP \ --disable_detector sv \ --min_tumor_allele_frac 3e-3 \ --min_tumor_lod 3.0 \ --min_init_tumor_lod 1.0 \ --assemble_mode 4 \ --pcr_indel_model NONE \ --min_base_qual 40 \ --resample_depth 100000 \ output_tnscope.pre_filter.vcf.gz || \ { echo "TNscope failed"; exit 1; } # ****************************************** # 4. Variant filtration # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon pyexec $TNSCOPE_FILTER \ -v output_tnscope.pre_filter.vcf.gz \ --tumor_sample $TUMOR_SM \ -x ctdna --min_tumor_af 0.005 --min_depth 400 \ output_tnscope.filter.vcf.gz || \ { echo "TNscope filter failed"; exit 1; } ``` ### 4.5.6 TNscope®杂交捕获分析脚本 以下是TNscope使用配对正常样本对杂交捕获数据进行体细胞变异检测的脚本示例: ``` #!/bin/sh # Copyright (c) 2016-2024 Sentieon Inc. All rights reserved # ******************************************* # Script to perform TNscope variant calling on # hybridization captured tumor sample with # coverage over 200x and a matched normal sample. # ******************************************* set -eu # Update with the fullpath location of your sample fastq TUMOR_SM="tumor_sample" #sample name TUMOR_RGID="rg_$TUMOR_SM" #read group ID NORMAL_SM="normal_sample" #sample name NORMAL_RGID="rg_$NORMAL_SM" #read group ID PL="ILLUMINA" #or other sequencing platform FASTQ_FOLDER="/home/pipeline/samples" TUMOR_FASTQ_1="$FASTQ_FOLDER/tumor_1.fastq.gz" TUMOR_FASTQ_2="$FASTQ_FOLDER/tumor_2.fastq.gz" #If using Illumina paired data NORMAL_FASTQ_1="$FASTQ_FOLDER/normal_1.fastq.gz" NORMAL_FASTQ_2="$FASTQ_FOLDER/normal_2.fastq.gz" #If using Illumina paired data # Update with the location of the reference data files FASTA_DIR="/home/regression/references/b37" FASTA="$FASTA_DIR/human_g1k_v37_decoy.fasta" KNOWN_DBSNP="$FASTA_DIR/dbsnp_138.b37.vcf.gz" INTERVAL_FILE="targeted_regions.bed" # Update with the location of the Sentieon software package and license file SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-|release_version| TNSCOPE_FILTER=/home/sentieon-scripts/tnscope_filter/tnscope_filter.py export SENTIEON_LICENSE=/home/Licenses/Sentieon.lic #or using licsrvr: c1n11.sentieon.com:5443 # Other settings NT=$(nproc) #number of threads to use in computation, set to number of cores in the server START_DIR="$PWD/test/TNscope" #Determine where the output files will be stored # ************************************************************************************************************************************************************************ # ****************************************** # 0. Setup # ****************************************** WORKDIR="$START_DIR" mkdir -p $WORKDIR LOGFILE=$WORKDIR/run.log exec >$LOGFILE 2>&1 cd $WORKDIR # ****************************************** # 1a. Mapping reads with BWA-MEM, sorting for the tumor sample # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$TUMOR_RGID\tSM:$TUMOR_SM\tPL:$PL" \ -t $NT -K 10000000 $FASTA $TUMOR_FASTQ_1 $TUMOR_FASTQ_2 || \ { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -o tumor_sorted.bam -t $NT --sam2bam -i - || \ { echo "Alignment1 failed"; exit 1; } # ****************************************** # 1b. Mapping reads with BWA-MEM, sorting for the normal sample # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$NORMAL_RGID\tSM:$NORMAL_SM\tPL:$PL" \ -t $NT -K 10000000 $FASTA $NORMAL_FASTQ_1 $NORMAL_FASTQ_2 || \ { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -o normal_sorted.bam -t $NT --sam2bam -i - || \ { echo "Alignment2 failed"; exit 1; } # ****************************************** # 2a. Metrics for the tumor sample # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_sorted.bam \ --algo MeanQualityByCycle tumor_mq_metrics.txt \ --algo QualDistribution tumor_qd_metrics.txt --algo GCBias \ --summary tumor_gc_summary.txt tumor_gc_metrics.txt --algo AlignmentStat \ --adapter_seq '' tumor_aln_metrics.txt \ --algo InsertSizeMetricAlgo tumor_is_metrics.txt \ --algo CoverageMetrics --omit_base_output tumor_coverage_metrics || \ { echo "Metrics1 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o tumor_gc-report.pdf tumor_gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution \ -o tumor_qd-report.pdf tumor_qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle \ -o tumor_mq-report.pdf tumor_mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo \ -o tumor_is-report.pdf tumor_is_metrics.txt # ****************************************** # 2b. Metrics for the normal sample # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i normal_sorted.bam \ --algo MeanQualityByCycle normal_mq_metrics.txt \ --algo QualDistribution normal_qd_metrics.txt --algo GCBias \ --summary normal_gc_summary.txt normal_gc_metrics.txt --algo AlignmentStat \ --adapter_seq '' normal_aln_metrics.txt \ --algo InsertSizeMetricAlgo normal_is_metrics.txt \ --algo CoverageMetrics --omit_base_output normal_coverage_metrics || \ { echo "Metrics2 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o normal_gc-report.pdf normal_gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution \ -o normal_qd-report.pdf normal_qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle \ -o normal_mq-report.pdf normal_mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo \ -o normal_is-report.pdf normal_is_metrics.txt # ****************************************** # 3a. Remove duplicate reads for the tumor sample. # It is possible to remove instead of mark # duplicates by adding the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i tumor_sorted.bam --algo LocusCollector \ --fun score_info tumor_score.txt || { echo "LocusCollector1 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i tumor_sorted.bam --algo Dedup \ --score_info tumor_score.txt --metrics tumor_dedup_metrics.txt tumor_deduped.bam || \ { echo "Dedup1 failed"; exit 1; } # ****************************************** # 3b. Remove duplicate reads for the normal sample. # It is possible to remove instead of mark # duplicates by adding the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i normal_sorted.bam --algo LocusCollector \ --fun score_info normal_score.txt || { echo "LocusCollector2 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i normal_sorted.bam --algo Dedup \ --score_info normal_score.txt --metrics normal_dedup_metrics.txt normal_deduped.bam || \ { echo "Dedup2 failed"; exit 1; } # ****************************************** # 4. Somatic and Structural variant calling # ****************************************** # Consider adding `--disable_detector sv` if not interested in SV calling $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT \ ${INTERVAL_FILE:+--interval $INTERVAL_FILE} \ -i tumor_deduped.bam -i normal_deduped.bam \ --algo TNscope \ --tumor_sample $TUMOR_SM --normal_sample $NORMAL_SM \ --dbsnp $KNOWN_DBSNP \ --min_tumor_allele_frac 0.009 \ --max_fisher_pv_active 0.05 \ --max_normal_alt_cnt 10 \ --max_normal_alt_frac 0.01 \ --max_normal_alt_qsum 250 \ --sv_mask_ext 10 \ --prune_factor 0 \ --assemble_mode 2 \ output_tnscope.pre_filter.vcf.gz || \ { echo "TNscope failed"; exit 1; } # ****************************************** # 5. Variant filtration # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon pyexec $TNSCOPE_FILTER \ -v output_tnscope.pre_filter.vcf.gz \ --tumor_sample $TUMOR_SM \ -x tissue_panel --min_tumor_af 0.0095 --min_depth 200 \ output_tnscope.filter.vcf.gz || \ { echo "TNscope filter failed"; exit 1; } ``` ### 4.5.7 TNscope® 全基因组分析脚本 以下是TNscope在全基因组测序中适用于测序深度达到50x的样本的脚本示例: ``` #!/bin/sh # Copyright (c) 2016-2024 Sentieon Inc. All rights reserved # ******************************************* # Script to perform TNscope variant calling on # samples up to 50x coverage using a matched # paired Tumor+normal sample with fastq files # named normal_1.fastq.gz, normal_2.fastq.gz # tumor_1.fastq.gz, tumor_2.fastq.gz # ******************************************* set -eu # Update with the fullpath location of your sample fastq TUMOR_SM="tumor_sample" #sample name TUMOR_RGID="rg_$TUMOR_SM" #read group ID NORMAL_SM="normal_sample" #sample name NORMAL_RGID="rg_$NORMAL_SM" #read group ID PL="ILLUMINA" #or other sequencing platform FASTQ_FOLDER="/home/pipeline/samples" TUMOR_FASTQ_1="$FASTQ_FOLDER/tumor_1.fastq.gz" TUMOR_FASTQ_2="$FASTQ_FOLDER/tumor_2.fastq.gz" #If using Illumina paired data NORMAL_FASTQ_1="$FASTQ_FOLDER/normal_1.fastq.gz" NORMAL_FASTQ_2="$FASTQ_FOLDER/normal_2.fastq.gz" #If using Illumina paired data # Update with the location of the reference data files FASTA_DIR="/home/regression/references/b37/" FASTA="$FASTA_DIR/human_g1k_v37_decoy.fasta" KNOWN_DBSNP="$FASTA_DIR/dbsnp_138.b37.vcf.gz" PON= # the TNscope panel-of-normals VCF # Update with the location of the Sentieon software package and license file SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-|release_version| export SENTIEON_LICENSE=/home/Licenses/Sentieon.lic #or using licsrvr: c1n11.sentieon.com:5443 # Other settings NT=$(nproc) #number of threads to use in computation, set to number of cores in the server START_DIR="$PWD/test/TNscope" #Determine where the output files will be stored BCFTOOLS_BINARY="/home/release/other_tools/bcftools-1.7/bcftools" #bcftools >=1.7 is recommended MIN_QUAL=10 # You do not need to modify any of the lines below unless you want to tweak the pipeline # ************************************************************************************************************************************************************************ # ****************************************** # 0. Setup # ****************************************** WORKDIR="$START_DIR" mkdir -p $WORKDIR LOGFILE=$WORKDIR/run.log exec >$LOGFILE 2>&1 cd $WORKDIR # ****************************************** # 1a. Mapping reads with BWA-MEM, sorting for tumor sample # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$TUMOR_RGID\tSM:$TUMOR_SM\tPL:$PL" \ -t $NT -K 10000000 $FASTA $TUMOR_FASTQ_1 $TUMOR_FASTQ_2 || \ { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -o tumor_sorted.bam -t $NT --sam2bam -i - || \ { echo "Alignment1 failed"; exit 1; } # ****************************************** # 1b. Mapping reads with BWA-MEM, sorting for normal sample # ****************************************** #The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000 ( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -R "@RG\tID:$NORMAL_RGID\tSM:$NORMAL_SM\tPL:$PL" \ -t $NT -K 10000000 $FASTA $NORMAL_FASTQ_1 $NORMAL_FASTQ_2 || \ { echo -n 'BWA error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -o normal_sorted.bam -t $NT --sam2bam -i - || \ { echo "Alignment2 failed"; exit 1; } # ****************************************** # 2a. Metrics for tumor sample # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_sorted.bam \ --algo MeanQualityByCycle tumor_mq_metrics.txt \ --algo QualDistribution tumor_qd_metrics.txt --algo GCBias \ --summary tumor_gc_summary.txt tumor_gc_metrics.txt --algo AlignmentStat \ --adapter_seq '' tumor_aln_metrics.txt \ --algo InsertSizeMetricAlgo tumor_is_metrics.txt \ --algo CoverageMetrics tumor_coverage_metrics || \ { echo "Metrics1 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o tumor_gc-report.pdf tumor_gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution \ -o tumor_qd-report.pdf tumor_qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle \ -o tumor_mq-report.pdf tumor_mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo \ -o tumor_is-report.pdf tumor_is_metrics.txt # ****************************************** # 2b. Metrics for normal sample # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i normal_sorted.bam \ --algo MeanQualityByCycle normal_mq_metrics.txt \ --algo QualDistribution normal_qd_metrics.txt --algo GCBias \ --summary normal_gc_summary.txt normal_gc_metrics.txt --algo AlignmentStat \ --adapter_seq '' normal_aln_metrics.txt \ --algo InsertSizeMetricAlgo normal_is_metrics.txt \ --algo CoverageMetrics normal_coverage_metrics || \ { echo "Metrics2 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o normal_gc-report.pdf normal_gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution \ -o normal_qd-report.pdf normal_qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle \ -o normal_mq-report.pdf normal_mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo \ -o normal_is-report.pdf normal_is_metrics.txt # ****************************************** # 3a. Remove Duplicate Reads for tumor # sample. It is possible # to remove instead of mark duplicates # by adding the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i tumor_sorted.bam --algo LocusCollector \ --fun score_info tumor_score.txt || { echo "LocusCollector1 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i tumor_sorted.bam --algo Dedup \ --score_info tumor_score.txt --metrics tumor_dedup_metrics.txt tumor_deduped.bam || \ { echo "Dedup1 failed"; exit 1; } # ****************************************** # 3b. Remove Duplicate Reads for normal # sample. It is possible # to remove instead of mark duplicates # by adding the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i normal_sorted.bam --algo LocusCollector \ --fun score_info normal_score.txt || { echo "LocusCollector2 failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i normal_sorted.bam --algo Dedup \ --score_info normal_score.txt --metrics normal_dedup_metrics.txt normal_deduped.bam || \ { echo "Dedup2 failed"; exit 1; } # ****************************************** # 4. Somatic and Structural variant calling # ****************************************** # Consider adding `--disable_detector sv --trim_soft_clip` if not interested in SV calling $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i tumor_deduped.bam \ -i normal_deduped.bam \ --algo TNscope --tumor_sample $TUMOR_SM --normal_sample $NORMAL_SM \ --dbsnp $KNOWN_DBSNP ${PON:+--pon $PON} output_tnscope.pre_filter.vcf.gz || \ { echo "TNscope failed"; exit 1; } # ****************************************** # 5. Variant filtration # ****************************************** ( $BCFTOOLS_BINARY annotate -x "FILTER/triallelic_site" output_tnscope.pre_filter.vcf.gz || \ { echo "VCF filtering failed"; exit 1; } ) | \ ( $BCFTOOLS_BINARY filter -m + -s "insignificant" -e "(PV>0.25 && PV2>0.25) || (INFO/STR == 1 && PV>0.05)" || \ { echo "VCF filtering failed"; exit 1; } ) | \ ( $BCFTOOLS_BINARY filter -m + -s "orientation_bias" -e "FMT/FOXOG[0] == 1" || \ { echo "VCF filtering failed"; exit 1; } ) | \ ( $BCFTOOLS_BINARY filter -m + -s "strand_bias" -e "SOR > 3" || \ { echo "VCF filtering failed"; exit 1; } ) | \ ( $BCFTOOLS_BINARY filter -m + -s "low_qual" -e "QUAL < $MIN_QUAL" || \ { echo "VCF filtering failed"; exit 1; } ) | \ ( $BCFTOOLS_BINARY filter -m + -s "short_tandem_repeat" -e "RPA[0]>=10" || \ { echo "VCF filtering failed"; exit 1; } ) | \ ( $BCFTOOLS_BINARY filter -m + -s "noisy_region" -e "ECNT>5" || \ { echo "VCF filtering failed"; exit 1; } ) | \ ( $BCFTOOLS_BINARY filter -m + -s "read_pos_bias" -e "FMT/ReadPosRankSumPS[0] < -8" || \ { echo "VCF filtering failed"; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util vcfconvert - output_tnscope.filtered.vcf.gz || \ { echo "VCF filtering failed"; exit 1; } ``` ### 4.5.8 附录 #### (1) TNscope 命令行可选参数说明 - `--assemble_mode`:设置 assemble_mode=2 可以在准确性和速度之间取得良好平衡。而在处理更复杂的基因组区域时,可以使用assemble_mode=4。 - `--sv_mask_ext`:用于防止在结构变异断点附近进行 SNP/Indel 检测,以减少因比对紊乱导致的假阳性。 - `--max_fisher_pv_active`:开启并设置 P 值(PV)过滤阈值,该指标用于衡量特定位点在肿瘤样本与正常样本之间的统计学差异。 - `--min_tumor_allele_frac`:设置被视为潜在变异位点的最低肿瘤等位基因频率(AF)。 - `--min_init_tumor_lod`:初始变异检测阶段的最低肿瘤对数优势比。 - `--min_tumor_lod`:输出 VCF 文件中带有 TLOD 注释的最低肿瘤对数优势比过滤阈值。 - `--prune_factor`:设置 prune_factor=0 以开启自适应图剪枝算法。 #### (2)常见问题解答(FAQ) **1)AD 比率与输出 VCF 中报告的 AF 不匹配:** - 原因是两者的计算方式和定义不同,这是源于 GATK 的历史遗留问题。 - AD:基于输入 BAM 文件的原始堆叠计算,是在局部组装之前生成的。 - AF:是在局部组装完成后,支持参考等位基因(REF)与突变等位基因(ALT)读段数量的比率。 - 过滤机制不同:读段在参与这两项计算之前,会经过略有不同的过滤标准。 **2)使用正常样本对照库 (Panel of Normal, PoN):** - PoN 可以配合配对正常样本使用,也可以在无对照样本的情况下单独使用。 - 若变异位点出现在 PoN 中,会被标记上panel_of_normal 过滤器标签。 - 在没有配对正常样本时,PoN 非常有用。您可以参考“4.5.2 TNscope® 使用步骤“章节中的”(2)生成正常 VCF 面板文件”获取使用 TNscope 生成 PoN VCF 的说明。 **3)COSMIC 和 dbSNP 数据库文件的作用:** - 它们仅对“肿瘤-正常”配对样本模式产生影响,且仅影响过滤步骤。 - 这两个数据库仅用于确定判断胚系风险时的正常样本对数优势比阈值。 **4)复杂变异:** Sentieon 提供了一个实验性的 Python 脚本,用于合并相同相位内的相邻变异。该脚本处于实验阶段,可在 https://github.com/Sentieon/sentieon-scripts/blob/master/merge_mnp/merge_mnp.py 中获取。 ## 4.6 RNAseq Sentieon® Genomics软件的一个典型用途是执行Broad研究所最佳实践中推荐的RNA分析生信分析流程,该流程描述在 https://gatk.broadinstitute.org/hc/en-us/articles/360035531192-RNAseq-short-variant-discovery-SNPs-Indels- 。 ### 4.6.1 概述 在这个生信分析流程中,您需要以下输入文件: - FASTA 文件:包含与待分析样本对应的参考基因组核苷酸序列。 - FASTQ 文件:一个或多个包含待分析样本核苷酸序列的文件。这些文件包含了来自 DNA 测序(此处指从 RNA 反转录而来的 cDNA 测序)的原始读段。软件支持输入经过 GZIP 压缩的 FASTQ 文件。软件仅支持包含 Sanger 格式(Phred+33)质量分数的文件。 - (可选)dbSNP 数据库:您希望包含在流程中的单核苷酸多态性数据库数据,以 VCF 文件形式使用;您可以使用经过 bgzip 压缩并建立索引的 VCF 文件。 - (可选)已知位点集合:您希望包含在流程中的多个已知位点集合,以 VCF 文件形式使用;您可以使用经过 bgzip 压缩并建立索引的 VCF 文件。 典型的生信分析流程包括以下步骤: 1. 将读数映射到参考基因组:此步骤将 FASTQ 文件中的读段比对并映射到 FASTA 文件中的参考基因组上。该步骤确保了数据能够被置于基因组上下文背景中(确定其位置)。 3. 去除或标记重复序列:此步骤检测指示同一 RNA 分子被多次测序的读段。这些重复序列不具信息性,不应作为额外的支持证据进行计数。 4. 在剪接位点切分读段:此步骤通过去除 N 碱基区域将 RNA 读段切分为外显子片段,同时保持分组信息,并硬切除任何伸入内含子区域的序列。此外,该步骤还会通过将质量值从 255 转换为 60,重新分配来自 STAR 的映射质量值,使其与后续步骤的预期保持一致。 5. (可选)碱基质量分数重校准(BQSR):此步骤修正序列读段数据中单个碱基的质量分数。此操作可消除由测序方法学产生的实验偏好性。 6. 变异检测:此步骤识别您的数据相对于参考基因组显示变异的位点,并计算每个样本在该位点的基因型。 ### 4.6.2 RNAseq使用步骤 #### (1)将读数映射到参考基因组 运行单条命令以高效地使用STAR执行对齐,并使用Sentieon®软件创建BAM文件并进行排序: ``` sentieon STAR --runThreadN NUMBER_THREADS --genomeDir STAR_REFERENCE \ --readFilesIn FASTQ FASTQ2 --readFilesCommand "zcat" \ --outStd BAM_Unsorted --outSAMtype BAM Unsorted --outBAMcompression 0 \ --outSAMattrRGline ID:GROUP_NAME SM:SAMPLE_NAME PL:PLATFORM \ --twopassMode Basic --twopass1readsN -1 --sjdbOverhang READ_LENGTH_MINUS_1 \ | sentieon util sort -r REFERENCE -o SORTED_BAM -t NUMBER_THREADS -i - ``` STAR的输入和选项在手册中有描述。 该命令需要以下输入参数: - `NUMBER_THREADS`:计算中使用的线程数。我们建议该数量不要超过系统中可用的计算核心数。我们建议STAR和util二进制程序使用相同数量的线程。 - `STAR_REFERENCE`:参考 FASTA 文件对应的 STAR 基因组索引目录位置。请确保 STAR 所需的所有必要参考数据均位于同一位置且命名一致。 - `REFERENCE`:参考 FASTA 文件的路径及文件名。请确保该 FASTA 文件及其对应的 FAI 索引文件与 `STAR_REFERENCE` 索引目录中的构建版本和命名规范相匹配。 - `FASTQ`:样本FASTQ文件的路径。如果数据来自双端测序技术,您还需要输入FASTQ2作为相应的配对样本FASTQ文件。您需要确保`--readFilesCommand`选项中使用的程序与输入FASTQ的压缩状态相匹配,即:如果FASTQ文件被压缩,您应使用`zcat`,而如果FASTQ文件未被压缩,您应使用`cat`。 - `GROUP_NAME`: 将添加到读组头行中的读组标识符。在您计划使用的所有数据集中,`RG:ID` 必须是唯一的。 - `SAMPLE_NAME`:将添加到读组头行中的样本名称。 - `PLATFORM`:用于测序DNA的测序平台名称。可能的选项有:ILLUMINA(当fastq文件在Illumina™机器上产生时);IONTORRENT(当fastq文件在Life Technologies™ Ion-Torrent™机器上产生时);ELEMENT(当fastq文件在Element Biosciences™机器上产生时);DNBSEQ(当fastq文件在MGI™机器上产生时);ULTIMA(当fastq文件在Ultima Genomics™机器上产生时)。 - `READ_LENGTH_MINUS_1`:输入数据的读长减去 1 的数值。 - `SORTED_BAM`:排序后的映射BAM输出文件的路径和文件名。系统将创建相应的索引文件(.bai)。 #### (2)去除或标记重复 在比对和排序BAM文件后,需要运行两条独立的命令来移除或标记重复。第一条命令收集读段信息,第二条命令执行去重复操作;`--rmdup` 选项控制是直接移除重复读段还是仅将其标记为重复。 ``` sentieon driver -t NUMBER_THREADS -i SORTED_BAM \ --algo LocusCollector --rna [--consensus] [--umi_tag XR] --fun score_info SCORE.gz sentieon driver -t NUMBER_THREADS -i SORTED_BAM \ --algo Dedup [--rmdup] --score_info SCORE.gz DEDUPED_BAM ``` 这些命令需要以下输入参数: - `NUMBER_THREADS`:计算中将使用的线程数。建议该数量不要超过系统中可用的计算核心数。 - `SORTED_BAM`:前一个映射阶段存储结果的路径。 - `SCORE.gz`:临时得分输出文件的路径和文件名。确保两条命令使用的是用一个文件。 - `DEDUPED_BAM`:去重复BAM输出文件的路径和文件名。系统将创建相应的索引文件(.bai)。 #### (3)在剪接位点切分读段 运行单条命令来执行读段向外显子片段的切分,并重新分配来自 STAR 的映射质量值。 ``` sentieon driver -t NUMBER_THREADS -r REFERENCE -i DEDUPED_BAM \ --algo RNASplitReadsAtJunction --reassign_mapq 255:60 SPLIT_BAM ``` 该命令需要以下输入参数: - `NUMBER_THREADS`:计算中将使用的线程数。建议该数量不要超过系统中可用的计算核心数。 - `REFERENCE`:参考FASTA文件的路径。需确保参考序列与映射阶段使用的完全一致。 - `DEDUPED_BAM`:前一个去重阶段存储结果的路径。 - `SPLIT_BAM`:包含切分后读段的 BAM 输出文件的路径及文件名。系统将创建对应的索引文件(.bai)。 #### (4)碱基质量分数重校准(BQSR;可选) 碱基质量分数重校准(BQSR)阶段的用法与DNAseq阶段相同;对于此阶段,您可以参考 *第4.1.2节* 获取详细的使用说明。 #### (5)RNA变异检测 运行单条命令进行变异检测,并同时应用此前计算得到的 BQSR。RNA 变异检测可以使用 Haplotyper 或DNAscope算法。在命令中,您应当使用 `--trim_soft_clip` 选项,并设置比 DNAseq® 变异检测更低的 Phred 格式最小置信度阈值。这意味着您应当将 `call_conf` 和 `emit_conf` 均设置为 20,而非默认的 30。 ``` sentieon driver -t NUMBER_THREADS -r REFERENCE -i SPLIT_BAM \ [-q RECAL_DATA.TABLE] --algo Haplotyper --trim_soft_clip \ --call_conf 20 --emit_conf 20 [-d dbSNP] VARIANT_VCF ``` 如果您想使用DNAscope进行检测,命令将是: ``` sentieon driver -t NUMBER_THREADS -r REFERENCE -i SPLIT_BAM \ [-q RECAL_DATA.TABLE] --algo DNAscope --trim_soft_clip \ --call_conf 20 --emit_conf 20 [-d dbSNP] VARIANT_VCF ``` 该命令需要以下输入参数: - `NUMBER_THREADS`:计算中将使用的线程数。建议该数量不要超过系统中可用的计算核心数。 - `REFERENCE`:参考FASTA文件的路径。需确保参考序列与映射阶段使用的完全一致。 - `SPLIT_BAM`:前一个RNASplitReadsAtJunction阶段存储结果的路径。 - `VARIANT_VCF`:变异检测输出文件的路径和文件名。系统将创建相应的索引文件。如果使用 `.gz` 扩展名,工具将输出压缩文件。 该命令的以下输入参数是可选的: - `RECAL_DATA.TABLE`:前一个BQSR阶段存储结果的路径。 - `dbSNP`:将用于标记已知变异的单核苷酸多态性数据库(dbSNP)的路径和文件名。您只能使用一个dbSNP文件。 ### 4.6.3 RNAseq标准分析脚本 以下是Sentieon RNAseq标准的分析脚本示例: ``` #!/bin/sh # Copyright (c) 2016-2020 Sentieon Inc. All rights reserved # ******************************************* # Script to perform RNA variant calling # using a single sample with fastq files # named 1.fastq.gz and 2.fastq.gz # ******************************************* set -eu # Update with the fullpath location of your sample fastq SM="sample" #sample name RGID="rg_$SM" #read group ID PL="ILLUMINA" #or other sequencing platform READ_LENGTH=101 #the read length for the sample FASTQ_FOLDER="/home/pipeline/samples" FASTQ_1="$FASTQ_FOLDER/rna1.fastq.gz" FASTQ_2="$FASTQ_FOLDER/rna2.fastq.gz" #If using Illumina paired data # Update with the location of the reference data files FASTA_DIR="/home/regression/references/b37/" FASTA="$FASTA_DIR/human_g1k_v37_decoy.fasta" KNOWN_DBSNP="$FASTA_DIR/dbsnp_138.b37.vcf.gz" KNOWN_INDELS="$FASTA_DIR/1000G_phase1.indels.b37.vcf.gz" KNOWN_MILLS="$FASTA_DIR/Mills_and_1000G_gold_standard.indels.b37.vcf.gz" #comment if STAR should generate a new genomeDir STAR_FASTA="$FASTA_DIR/human_g1k_v37_decoy.fasta.genomeDir" #uncomment if you would like to use a bed file #INTERVAL_FILE="$FASTA_DIR/TruSeq_exome_targeted_regions.b37.bed" # Update with the location of the Sentieon software package and license file SENTIEON_INSTALL_DIR=/home/release/sentieon-genomics-|release_version| export SENTIEON_LICENSE=/home/Licenses/Sentieon.lic #or using licsrvr: c1n11.sentieon.com:5443 # Other settings NT=$(nproc) #number of threads to use in computation, set to number of cores in the server START_DIR="$PWD/test/RNAseq" #Determine where the output files will be stored # You do not need to modify any of the lines below unless you want to tweak the pipeline # ************************************************************************************************************************************************************************ # ****************************************** # 0. Setup # ****************************************** WORKDIR="$START_DIR/${SM}" mkdir -p $WORKDIR LOGFILE=$WORKDIR/run.log exec >$LOGFILE 2>&1 cd $WORKDIR DRIVER_INTERVAL_OPTION="${INTERVAL_FILE:+--interval $INTERVAL_FILE}" # ****************************************** # 1. Mapping reads with STAR # ****************************************** if [ -z "$STAR_FASTA" ]; then STAR_FASTA="genomeDir" # The genomeDir generation could be reused mkdir $STAR_FASTA $SENTIEON_INSTALL_DIR/bin/sentieon STAR --runMode genomeGenerate \ --genomeDir $STAR_FASTA --genomeFastaFiles $FASTA --runThreadN $NT || \ { echo "STAR index failed"; exit 1; } fi #perform the actual alignment and sorting ( $SENTIEON_INSTALL_DIR/bin/sentieon STAR --twopassMode Basic --genomeDir $STAR_FASTA \ --runThreadN $NT --outStd BAM_Unsorted --outSAMtype BAM Unsorted \ --outBAMcompression 0 --twopass1readsN -1 --sjdbOverhang `expr "$READ_LENGTH" - 1` \ --readFilesIn $FASTQ_1 $FASTQ_2 --readFilesCommand "zcat" \ --outSAMattrRGline ID:$RGID SM:$SM PL:$PL || { echo -n 'STAR error'; exit 1; } ) | \ $SENTIEON_INSTALL_DIR/bin/sentieon util sort -r $FASTA -o sorted.bam \ -t $NT --bam_compression 1 -i - || { echo "STAR alignment failed"; exit 1; } # ****************************************** # 2. Metrics # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver $DRIVER_INTERVAL_OPTION -r $FASTA -t $NT \ -i sorted.bam --algo MeanQualityByCycle mq_metrics.txt --algo QualDistribution \ qd_metrics.txt --algo GCBias --summary gc_summary.txt gc_metrics.txt \ --algo AlignmentStat --adapter_seq '' aln_metrics.txt --algo InsertSizeMetricAlgo \ is_metrics.txt || { echo "Metrics failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o gc-report.pdf gc_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution -o qd-report.pdf qd_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle -o mq-report.pdf mq_metrics.txt $SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo -o is-report.pdf is_metrics.txt # ****************************************** # 3. Remove Duplicate Reads. It is possible # to remove instead of mark duplicates # by adding the --rmdup option in Dedup # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i sorted.bam --algo LocusCollector \ --fun score_info score.txt || { echo "LocusCollector failed"; exit 1; } $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT -i sorted.bam --algo Dedup \ --score_info score.txt --metrics dedup_metrics.txt deduped.bam || \ { echo "Dedup failed"; exit 1; } # ****************************************** # 2a. Coverage metrics # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i deduped.bam \ --algo CoverageMetrics coverage_metrics || { echo "CoverageMetrics failed"; exit 1; } # ****************************************** # 4. Split reads at Junction # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver -r $FASTA -t $NT -i deduped.bam \ --algo RNASplitReadsAtJunction --reassign_mapq 255:60 splitted.bam || \ { echo "RNASplitReadsAtJunction failed"; exit 1; } # ****************************************** # 6. Base recalibration # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver $DRIVER_INTERVAL_OPTION -r $FASTA -t $NT \ -i splitted.bam --algo QualCal -k $KNOWN_DBSNP -k $KNOWN_MILLS -k $KNOWN_INDELS \ recal_data.table $SENTIEON_INSTALL_DIR/bin/sentieon driver $DRIVER_INTERVAL_OPTION -r $FASTA -t $NT \ -i splitted.bam -q recal_data.table --algo QualCal -k $KNOWN_DBSNP -k $KNOWN_MILLS \ -k $KNOWN_INDELS recal_data.table.post $SENTIEON_INSTALL_DIR/bin/sentieon driver -t $NT --algo QualCal --plot \ --before recal_data.table --after recal_data.table.post recal.csv $SENTIEON_INSTALL_DIR/bin/sentieon plot QualCal -o recal_plots.pdf recal.csv # ****************************************** # 7. HC Variant caller for RNA # ****************************************** $SENTIEON_INSTALL_DIR/bin/sentieon driver $DRIVER_INTERVAL_OPTION -r $FASTA -t $NT \ -i splitted.bam -q recal_data.table --algo Haplotyper -d $KNOWN_DBSNP \ --trim_soft_clip --emit_conf=20 --call_conf=20 output-hc-rna.vcf.gz || \ { echo "Haplotyper failed"; exit 1; } ``` ## 4.7 LongReads SV ### 4.7.1 概述 本节描述了使用Sentieon® LongReads SV从PacBio HiFi和Oxford Nanopore长读段中检测结构变异(SV)。准确的长读长测序技术能够准确发现之前短读长测序无法获得的大型SV事件。 Sentieon® LongReads SV 能够充分利用极长的读段长度优势,为 PacBio HiFi 和 Oxford Nanopore 输入的长读段数据提供快速且准确的大型结构变异事件检测及基因分型。 Sentieon® LongReads SV 需要已比对的 BAM 或 CRAM 文件作为输入,并输出 VCF 格式的变异结果。我们建议使用 Sentieon® 加速版的 Minimap2 和排序工具,以实现高效且准确的比对。 目前,Sentieon® LongReads SV可检测和分型大型INDEL事件,并输出高置信度的一致INDEL碱基序列。未来版本将添加对其他SV事件类型的支持。 如有任何其他问题,请联系Sentieon® Inc.的技术持:support@insvast.com。 ### 4.7.2 使用LongReads SV 要运行Sentieon LongReads SV,请运行以下命令: ``` sentieon driver -t NUMBER_THREADS -r REFERENCE -i INPUT_BAM \ --algo LongReadSV [--min_sv_size MIN_SV_SIZE] [--min_map_qual MIN_MAP_QUAL]\ --model MODEL/longreadsv.model OUT_SV_VCF ``` 该命令需要以下参数: - `-t NUMBER_THREADS`:计算中将使用的线程数。建议该数量不要超过系统中可用的计算核心数。 - `-r REFERENCE`:参考FASTA文件的路径。需确保参考序列与映射阶段使用的完全一致。 - `-i INPUT_BAM`:输入比对文件,应为已建立索引的 BAM 或 CRAM 文件。该文件包含使用 `minimap2`(或针对 HiFi 数据,使用带有 `--preset HIFI` 参数的 `pbmm2`)比对后的 PacBio HiFi 或 Oxford Nanopore 读段。当使用 Sentieon minimap2 进行比对时,建议使用针对 HiFi 或 ONT 优化的 Sentieon 模型。如果使用原版 `minimap2`,建议设置 `-x map-hifi` 或 `-x map-ont` 选项,这将为输入的测序数据开启推荐的 `minimap2` 设置。 - `--model MODEL`:输入模型包文件,其中包含了针对 PacBio HiFi 或 Oxford Nanopore 长读段预设的编码配置。您需要选择与输入比对文件的测序平台相匹配的模型文件。 请参考Sentieon的 https://github.com/Sentieon/sentieon-models 页面下载最新的SV模型。 该命令需要以下位置参数: - `OUT_SV_VCF`:输出 SV 检测结果的路径及文件名,支持 `.vcf` 或 `.vcf.gz` 后缀。系统将同时创建一个对应的索引文件。如果使用 `.gz` 后缀,工具将输出压缩文件。 以下参数是可选的: - `--min_sv_size MIN_SV_SIZE`:输出结构变异的最小长度限制,单位为碱基对(默认值:40)。 - `--min_map_qual MIN_MAP_QUAL`:读段的最小映射质量(默认值:20)。 ### 4.7.3 评估LongReads SV结果 Sentieon® 推荐使用Sentieon® hap-eval 基于组装的单倍型对 SV 检测结果进行精确评估和对比。Hap-eval 是一款开源的、专为结构变异基准测试设计的 VCF 对比引擎。您可在 https://github.com/Sentieon/hap-eval 上下载。 ## 4.8 Sentieon CLI Sentieon-cli 通过单条命令即可实现 Sentieon® 流程的完整运行。设计初衷是显著提升复杂的多阶段比对及变异检测流程的易用性。 ### 4.8.1 Sentieon Cli安装说明 #### (1)从sdist安装(推荐) 从GitHub发布页面 https://github.com/sentieon/sentieon-cli/releases/ 下载最新的tar.gz文件,并使用`pip`安装软件包: ``` curl -LO https://github.com/sentieon/sentieon-cli/releases/download/v1.1.0/sentieon_cli-1.1.0.tar.gz pip install sentieon_cli-1.1.0.tar.gz ``` #### (2)使用Poetry安装 如果需要,请为项目创建一个新的Python虚拟环境: ``` # Create a new venv, if needed python3 -m venv /path/to/new/virtual/environment/sentieon_cli # Activate the venv source /path/to/new/virtual/environment/sentieon_cli/bin/activate ``` sentieon-cli使用poetry进行打包和依赖管理。首先,你需要安装poetry: ``` pip install poetry ``` 克隆此仓库并进入根目录: ``` git clone https://github.com/sentieon/sentieon-cli.git cd sentieon-cli ``` 使用poetry将sentieon-cli安装到虚拟环境中: ``` poetry install ``` 然后你可以从虚拟环境中运行命令: ``` sentieon-cli ... ``` #### (3)全局参数 sentieon-cli支持以下全局参数: - `--verbose`:输出详细日志记录。 - `--debug`:调试模式,输出更详尽的日志记录。 #### (4) 支持的流程 - DNAscope - 用于从短读段数据进行胚系SNV和indel检测的DNAscope流程实现。 - DNAscope LongRead - 用于从长读段数据进行胚系SNV和indel检测的DNAscope LongRead流程实现。 - DNAscope Hybrid - DNAscope 短读段-长读段混合分析流程。 - Pangenome - 利用泛基因组图谱进行短读段比对及变异检测的 DNAscope流程。 ### 4.8.2 DNAscope Sentieon® DNAscope 是一个专门针对短读段 DNA 测序数据设计的完整流程,涵盖了从序列比对到胚系细胞变异检测(包括 SNV、SV、Indel 和 CNV)的全过程。通过 sentieon-cli 工具,用户仅需一条命令即可运行完整的DNAscope 流程。该流程在兼顾了高准确性、高效率和易用性。 DNAscope 流程结合了贝叶斯统计方法与机器学习算法,以实现卓越的变异检测精度。它不仅支持全基因组测序,也支持靶向捕获(如全外显子组 WES)文库制备样本。 流程支持已比对的 BAM/CRAM格式输入,也支持未比对的 FASTQ、uBAM、uCRAM格式;流程最终输出 VCF(或 gVCF)格式的变异结果,以及 BAM/CRAM 格式的比对文件。 #### (1) 环境配置 **1)必备条件** - Sentieon® 软件包:202308 或更高版本。 - samtools:1.16 或更高版本,用于处理 uBAM/uCRAM 或重新比对。 - MultiQC:1.18 或更高版本,用于生成质控报告。 - 上述`sentieon`、`samtools` 和 `multiqc`可执行文件需添加至系统的 PATH 环境变量中。 **2)参考基因组** DNAscope 将相对于高质量的参考基因组序列来检测样本中存在的变异。除了参考基因组文件外,还需要提供 samtools fasta 索引文件(.fai)。序列比对还需要 bwa 索引文件。 我们建议比对到不含替代重叠群的参考基因组。如果基因组中存在替代重叠群,请同时提供一个“.alt”文件,以激活 bwa 中的Alt-aware 比对。 #### (2)使用说明 **1)从 FASTQ 进行比对和变异检测** 只需运行单条命令,即可完成从 FASTQ 文件进行的比对、预处理、 SNV、Indel 和 SV 的检测: ``` sentieon-cli dnascope [-h] \ -r REFERENCE \ --r1_fastq R1_FASTQ ... \ --r2_fastq R2_FASTQ ... \ --readgroups READGROUPS ... \ -m MODEL_BUNDLE \ [-d DBSNP] \ [-b BED] \ [--interval_padding INTERVAL_PADDING] \ [-t NUMBER_THREADS] \ [--pcr_free] \ [-g] \ [--duplicate_marking DUP_MARKING] \ [--assay ASSAY] \ [--consensus] \ [--dry_run] \ [--bam_format] \ sample.vcf.gz ``` 使用 FASTQ 输入时,DNAscope 流程需要以下必选参数: - `-r REFERENCE`: 参考基因组 FASTA 文件的位置。同时需要参考基因组索引文件(“.fai”)以及 bwa 比对索引文件。 - `--r1_fastq R1_FASTQ`: 输入的 R1 端 FASTQ 文件。可以多次使用该参数。若没有对应的 R2_FASTQ,则默认为单端数据。请注意,该流程为单样本处理,所有 FASTQ 文件必须来自同一个样本。 - `--r2_fastq R2_FASTQ`: 输入的 R2 端 FASTQ 文件。可以多次使用该参数。 - `--readgroups READGROUPS`: 每个 FASTQ 的读组信息。流程要求--r1_fastq 和 --readgroups 参数的数量必须一致。 - 示例参数:`--readgroups"@RG\tID:HG002-1\tSM:HG002\tLB:HG002-LB-1\tPL:ILLUMINA"` - `-m MODEL_BUNDLE`: 模型包的位置。模型文件可在 Sentieon 官方仓库中找到。 - `sample.vcf.gz`: 用于存储 SNV 和 Indel 检测结果的输出 VCF 文件位置。流程要求输出文件名必须以 .vcf.gz 为后缀。不带后缀的文件路径将用作其他输出文件的主文件名。 DNAscope 流程支持以下可选参数: - `-d DBSNP`: 单核苷酸多态性数据库(dbSNP)的位置,用于在 VCF 中标记已知变异。支持 VCF 或 bgzip 压缩的 VCF 格式。仅支持输入单个文件。提供此文件将通过 dbSNP 的 refSNP ID 编号对变异进行注释。需要提供VCF 索引文件。 - `-b BED`: 指定参考基因组上的区间以限制变异检测范围,格式为BED。提供此文件后,变异检测将仅限于 BED 文件内的区间。若未提供,软件将处理全基因组。 - `--interval_padding INTERVAL_PADDING`: 为输入区间边缘添加 `INTERVAL_PADDING` 个碱基的填充。默认值为 0。 - `-t NUMBER_THREADS`: 软件运行并行任务时使用的线程数。此参数可选;若省略,流程将默认使用服务器的所有可用线程。 - `--pcr_free`: 使用 `--pcr_indel_model NONE` 模式进行变异检测,适用于PCR-free(无 PCR 扩增)文库。系统仍会进行去重复以识别光学重复。 - `-g`: 在输出 VCF 文件的同时,输出 gVCF 格式的变异结果。工具将输出一个经过 bgzip 压缩的 gVCF 文件及其索引文件。 - `--duplicate_marking DUP_MARKING`: 重复序列标记设置。`markdup` 为标记重复读段;`rmdup` 为移除重复读段;`none` 为跳过重复序列标记。默认设置为 `markdup`。 - `--assay ASSAY`: 用于指标收集的实验类型设置,可选WGS(全基因组)或 WES(全外显子组)。默认设置为 WGS。 - `--consensus`: 在重复序列标记阶段生成共识读段。 - `-h`: 打印命令行帮助信息并退出。 - `--dry_run`: 打印流程命令,但不实际执行。 - `--bam_format`: 输出比对文件时使用 BAM 格式替代 CRAM 格式。 **2)从 uBAM 或 uCRAM 进行比对和变异检测** 只需运行单条命令,即可完成从 uBAM 或 uCRAM 文件进行的比对、预处理、以及SNV、Indel 和 SV 的变异检测: ``` sentieon-cli dnascope [-h] \ -r REFERENCE \ -i SAMPLE_INPUT ... \ --align \ [--input_ref INPUT_REF] \ -m MODEL_BUNDLE \ [-d DBSNP] \ [-b BED] \ [--interval_padding INTERVAL_PADDING] \ [-t NUMBER_THREADS] \ [--pcr_free] \ [-g] \ [--duplicate_marking DUP_MARKING] \ [--assay ASSAY] \ [--consensus] \ [--dry_run] \ [--bam_format] \ sample.vcf.gz ``` 使用 uBAM 或 uCRAM 作为输入时,DNAscope 流程需要以下新增参数: - `-i SAMPLE_INPUT`:uBAM 或 uCRAM 格式的输入样本文件。可以通过在 `-i` 参数后列出多个文件来提供。 - ` --align`:指示流程对比对输入读段。 DNAscope 流程支持以下新增可选参数: - `--input_ref INPUT_REF`:用于解码输入文件的参考基因组 FASTA 文件。在使用 uCRAM 输入时必须提供此参数。该文件可以与 `-r` 参数指定的参考基因组不同。 **3)从坐标排序的 BAM 或 CRAM 进行比对和变异检测** 只需运行单条命令,即可完成从(已完成坐标排序的)BAM 或 CRAM 文件进行比对、预处理以及 SNV、Indel 和 SV 的检测: ``` sentieon-cli dnascope [-h] \ -r REFERENCE \ -i SAMPLE_INPUT ... \ --collate_align \ [--input_ref INPUT_REF] \ -m MODEL_BUNDLE \ [-d DBSNP] \ [-b BED] \ [--interval_padding INTERVAL_PADDING] \ [-t NUMBER_THREADS] \ [--pcr_free] \ [-g] \ [--duplicate_marking DUP_MARKING] \ [--assay ASSAY] \ [--consensus] \ [--dry_run] \ [--bam_format] \ sample.vcf.gz ``` 使用 BAM 或 CRAM 作为输入时,DNAscope 流程需要以下新增参数: - `--collate_align`:指示流程先对输入读段进行整理,然后再进行比对。 **4)从已排序的 BAM 或 CRAM 进行变异检测** 只需运行单条命令,即可完成从(已排序的)BAM 或 CRAM 文件进行预处理,以及 SNV、Indel 和 SV 的检测: ``` sentieon-cli dnascope [-h] \ -r REFERENCE \ -i SAMPLE_INPUT ... \ -m MODEL_BUNDLE \ [-d DBSNP] \ [-b BED] \ [--interval_padding INTERVAL_PADDING] \ [-t NUMBER_THREADS] \ [--pcr_free] \ [-g] \ [--duplicate_marking DUP_MARKING] \ [--assay ASSAY] \ [--consensus] \ [--dry_run] \ [--bam_format] \ sample.vcf.gz ``` 如果不提供 `--align` 和 `--collate_align` 参数,流程将直接从输入的读段中检测变异。 #### (3)流程输出 **1)输出文件列表** 当 使 用 默 认 参 数 处 理 全 基 因 组 FASTQ 数 据 且 指 定 的 输 出 文 件 名 为`sample.vcf.gz` 时,会生成以下文件: - `sample.vcf.gz`:在 `-b BED` 文件定义的基因组区域内的 SNV 和 Indel 变异检测结果。 - `sample_deduped.cram` 或 `sample_deduped.bam`:由输入 FASTQ 生成的经比对、坐标排序及去重标记后的读段数据。 - `sample_svs.vcf.gz`:由 DNAscope 和 SVSolver 检测到的 SV 结果。 - `sample_metrics`:包含该样本所有 QC 指标的目录。 - `sample_metrics/coverage*`:处理样本的覆盖度指标(仅适用于 WGS 样本)。 - `sample_metrics/{sample}.txt.alignment_stat.txt`:来自 AlignmentStat算法的比对统计指标。 - `sample_metrics/{sample}.txt.base_distribution_by_cycle.txt `: 来自BaseDistributionByCycle 算法的逐循环碱基分布指标。 - `sample_metrics/{sample}.txt.dedup_metrics.txt`:来自 Dedup 算法的去重统计指标。 - `sample_metrics/{sample}.txt.gc_bias*`:来自 GCBias 算法的 GC 偏好指标(仅适用于 WGS 样本)。 - `sample_metrics/{sample}.txt.insert_size.txt`:来自 InsertSizeMetricAlgo 算法的文库插入片段大小指标。 - `sample_metrics/{sample}.txt.mean_qual_by_cycle.txt`:来自 MeanQualityByCycle 算法的逐循环平均质量值指标。 - `sample_metrics/{sample}.txt.qual_distribution.txt`:来自 QualDistribution 算法的质量值分布指标。 - `sample_metrics/{sample}.txt.wgs.txt`:来自 WgsMetricsAlgo 算法的全基因组统计指标(仅适用于 WGS 样本)。 - `sample_metrics/{sample}.txt.hybrid-selection.txt`:来自 HsMetricAlgo 算法的杂交捕获(如全外显子组)统计指标。 - `sample_metrics/multiqc_report.html`:由 MultiQC 汇总并生成的交互式 QC 指标报告。 [**点击此处跳转至Sentieon中文手册(下册)**](https://doc.insvast.com/doc/64/)
chsnp
2026年2月9日 10:29
转发
收藏文档
上一篇
下一篇
手机扫码
复制链接
手机扫一扫转发分享
复制链接
Markdown文件
Word文件
PDF文档
PDF文档(打印)
分享
链接
类型
密码
更新密码
有效期